
Abstract
The G protein-coupled receptor 55 (GPR55) is expressed in multiple tissues, and has been implicated in cancer pathogenesis, but little is known about its role in the migratory behavior of cancer cells, particularly breast cancer cells. In this study we first showed that GPR55 expression levels in 38 metastatic lymph nodes of breast cancer patients were profoundly elevated, and were positively associated in human breast cancer cells with their migratory ability. Moreover, the plasma levels of GPR55 endogenous agonist L-a-lysophosphatidylinositol (LPI) were significantly increased in breast cancer patients compared with healthy individuals. In human breast cancer LM-MCF-7 and MDA-MB-231 cells, treatment with LPI (2.5 μmol/L) significantly increased filopodia formation and resulted in cell migration, which could be blocked either by the GPR55 antagonist CID16020046 or by siRNA-mediated GPR55 knockdown. Furthermore, dual-luciferase report gene assays showed that GPR55 upregulated HBXIP at the promoter; GPR55 expression levels were positively correlated with HBXIP expression levels in breast cancer tissues and 8 breast cancer cell lines. We also showed that the LPI/GPR55 axis promoted the migration of breast cancer cells via two mutually exclusive pathways — the HBXIP/p-ERK1/2/Capn4 and MLCK/MLC signaling pathways. In xenograft nude mouse model, loss of GPR55 mainly affected breast cancer cell metastasis and the formation of metastatic foci. Thus, GPR55 is involved in the migratory behavior of human breast cancer cells and could serve as a pharmacological target for preventing metastasis.
Similar content being viewed by others


Introduction
The G protein-coupled receptor 55 (GPR55) is a GPCR that is thought to be part of the endocannabinoid system (CB1 and CB2) but whose pharmacology is still under investigation. GPR55 mRNA has been detected in multiple tissues of the body, including the brain, spleen, and bones, and in cells such as adipocytes, gastrointestinal tract cells and islets of Langerhans1. Consequently, numerous studies have revealed various physiological functions of this receptor. Several reports have highlighted the role of GPR55 in several cancer types, such as glioma, melanoma and pancreatic cancer cells1,2,3,4. GPR55 couples to the G12/13 and Gq alpha subunits of heterotrimeric G protein complexes. Various studies have described the role of G12/13 signaling in promoting cancer cell proliferation, invasion, metastatic spread and angiogenesis5,6. Another study demonstrated that downregulation of GPR55 in a xenograft mouse model of glioblastoma reduces tumor growth and that GPR55 knockout mice are more resistant to skin carcinogenesis1,4. However, little is known regarding the role of GPR55 in the migratory behavior of cancer cells, particularly in breast cancer.
Although the specific pharmacology of GPR55 remains controversial, most evidence has indicated that GPR55 is activated by certain cannabinoid ligands and by the lysophospholipid L-a-lysophosphatidylinositol (LPI), as first discovered by Oka et al7. Moreover, LPI was observed to activate GPR55 in cells that endogenously express this receptor (eg, large dorsal root ganglion neurons8, osteoclasts9 and lymphoblastoid cells10), supporting the notion that this phospholipid is an endogenous GPR55 ligand. LPI is generated by hydrolyzing phosphatidylinositol via calcium-dependent phospholipase A2 and calcium-independent phospholipase A111. LPI is involved in numerous physiological processes, including reproduction, angiogenesis, apoptosis, and inflammation, all of which are closely associated with adipose tissue biology. Various lines of evidence have recently indicated a role of the endogenous LPI in cancer. Malignant ascites contain high levels of lysophospholipids—LPI levels in ascites collected from malignant ovarian samples are approximately 16-fold higher than those in ascites from non-malignant samples12. Furthermore, plasma LPI levels are augmented in patients with late-stage or recurring ovarian cancer13. In vitro studies have demonstrated that the malignant transformation of epithelial thyroid cells and fibroblasts using the oncogene Ras leads to the synthesis and release of LPI and that LPI induced the proliferation of these cells14. Oka et al found that LPI, which significantly increases cell proliferation, induces the rapid phosphorylation of extracellular signal-regulated kinase (ERK) and an increase in intracellular Ca2+ levels in cells expressing GPR557. However, the LPI levels in breast cancer patients and the role of LPI in regulating breast cancer cell migration require further investigation.
Mammalian hepatitis B X-interacting protein (HBXIP) is a 18-kDa protein that was originally identified by its interaction with the hepatitis B virus X protein, and its sequence is well conserved among mammalian species. HBXIP suppresses apoptosis in hepatoma cells and regulates centrosome duplication in HeLa cells15,16. Our group reported that HBXIP also plays crucial roles in promoting the proliferation and migration of breast cancer cells by acting as a transcription factor coactivator17,18,19,20. However, the mechanism by which HBXIP enhances breast cancer cell migration is poorly understood.
In this study, we identified that the LPI/GPR55 axis accelerated the migration of breast cancer cells. Interestingly, GPR55 was highly expressed in tissue samples from metastatic lymph nodes, and LPI expression was significantly increased in the plasma of breast cancer patients compared to healthy individuals. The LPI/GPR55 axis increased filopodia formation, thus resulting in cell migration. GPR55 upregulated HBXIP and Capn4 expression in breast cancer cells at the promoter, mRNA and protein levels. Moreover, the LPI/GPR55 axis acted through two mutually exclusive pathways—the HBXIP-p-ERK1/2/Capn4 and MLCK/MLC pathways—to promote cell migration. Loss of GPR55 primarily affected breast cancer cell metastasis and the formation of metastatic foci. Thus, our findings provide new insight into the mechanism of breast cancer cell migration.
Materials and methods
Cell culture, drugs, plasmids and siRNA
SK-BR3, HBL-100, 184A1, Hs578T, T47D and BT-549 cell lines were purchased from ATCC and cultured according to ATCC protocol. MCF-7, LM-MCF-7 (a highly metastatic subclone of the MCF-7 cell line21), and MDA-MB-231 cells were cultured in RPMI-1640 medium (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal calf serum, 2 mmol/L L-glutamine, and 100 U/mL penicillin/streptomycin and incubated in a humidified atmosphere at 37 °C containing 5% CO2. No passage higher than seven was used in experiments.
CID16020046 (4-[4-(3-hydroxyphenyl)-3-(4-methylphenyl)-6-oxo-1H,4H,5H,6H-pyrrolo [3,4-c] pyrazol-5-yl] benzoic acid) was obtained from Tocris Bioscience (Bristol, UK) and diluted in DMSO. LPI (L-α-lysophosphatidylinositol; Sigma, St Louis, MO, USA) was dissolved in deionized sterile water. Cells were pretreated for 1 h with 30 μmo/L PD98059 (Sigma, St Louis, MO, USA), 20 μmol/L ML-7 (Sigma, St Louis, MO, USA) or 2 μmol/L CID16020046 before subjecting to the following assays.
Human GPR55 (GenBank™ accession number NM_005683.3) total RNA was extracted from LM-MCF-7 cells and reverse transcribed into cDNA. Luciferase reporter vectors containing HBXIP and Capn4 promoters, HBXIP siRNA and control siRNA were used as described in our previous study22,23. The following siRNAs were used in this study: siGPR55: 5′-GGUUCUUGGCCAUCCGUUAtt-3′ and siControl: 5′-AAUGGUCAUGGUCUUAUUCC-3′. One day before transfection, cells (1×105 cells per well) were collected and seeded into a 6-well culture plate (n=3 per condition). Cells were transfected with the corresponding plasmids or siRNAs using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions.
Immunohistochemistry
A breast cancer tissue array (No 08C14) comprising duplicates of 49 samples of infiltrating primary carcinoma and 38 metastatic tumors was purchased from Xi-an Aomei Biotechnology (Xi-an, China). An immunohistochemical assay was performed as previously described17. The slides were incubated with anti-GPR55 (ab 203663, Abcam, China) antibodies at 4 °C overnight followed by 30 min of incubation at room temperature with biotinylated secondary antibodies and subsequent treatment with a streptavidin-peroxidase complex at room temperature for 30 min. Staining was developed using chromogen 3,3′-diaminobenzidine (DAB), and the slides were counterstained with Mayer's hematoxylin.
Human blood samples
A total of 50 blood samples were collected from 36 breast cancer patients and 14 healthy individuals (all provided written informed consent) at the Fourth Hospital of Hebei Medical University (Shijiazhuang, China). Blood samples were drawn into plasma separation tubes containing heparin and centrifuged within 2 h of collection at 1600×g for 10 min. The plasma was then transferred into cryotubes without disturbing the buffy coat layer. Ethical approval for this study was granted by the ethics committee of the Fourth Hospital of Hebei Medical University.
LC-MS of LPI
Lipid extraction was performed using 150 μL of plasma in the presence of 10 μL LPI 17:1 (100 μmol/L) as an internal standard, as described by Matyash et al24. LC-MS measurements for lipid quantification were performed as previously described by Fauland et al25 with slight modifications. LC was performed on a Kinetex reversed-phase C18 2.1 mm×150 mm (2.6 μm) column (Phenomenex, Torrance, CA, USA). Mobile phase A was 10 mmol/L ammonium acetate containing 0.1% formic acid. Mobile phase B was acetonitrile/2-propanol 5:2 (v/v) containing 10 mmol/L ammonium acetate and 0.1% formic acid. The binary gradient started with 35% to 70% B for 4 min and was then increased to 100% B within 16 min and further held for 10 min. The flow rate was 350 μL/min, the oven temperature was 50 °C, and the tray temperature was 10 °C. Ten-microliter plasma samples were injected for analysis. After each run, the column was flushed for 5 min with 35% B before the next run was started. An LTQ Orbitrap XL mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) was used for the analysis. Parallel MS/MS spectra were obtained in linear ion trap in preview mode, while the Orbitrap analyzer was run in full scan mode at a resolution of 100 000 (m/z 400) and in negative ESI mode from m/z 490 to 1100. Helium gas was used for the linear ion trap collision-induced dissociation spectra. Based on the LTQ-FT preview scan, the 3 most abundant ions were selected during data-dependent acquisition (DDA), fragmented in the linear ion trap analyzer and ejected at a nominal mass resolution.
Reverse transcription-PCR (RT-PCR) and real-time PCR
A total of 36 samples of breast cancer tissues were collected from patients (patient information is listed in Supplementary Table S1) undergoing breast cancer resection at the Fourth Hospital of Hebei Medical University (Shijiazhuang, China). Total RNA from cells was prepared using TRIzol (Invitrogen, Carlsbad, CA, USA). First-strand cDNA was synthesized using PrimeScript reverse transcriptase (TaKaRa Bio, Dalian, China) and an oligo-(dT) primer according to the manufacturer's instructions. The primers used to test HBXIP expression were previously reported26. The other primers used in this study were as follows: human GPR55, 5′-CCTCCCATTCAAGATGGTCC-3′ (sense) and 5′-GACGCTTCCGTACATGCTGA-3′ (antisense); GAPDH, 5′-CATCACCATCTTCCAGGAGCG-3′ (sense) and 5′-TGACCTTGCCCACAGCCTTG-3′ (anti-sense).
Wound healing assay
To determine the effect of the GPR55/LPI axis on cell migration, LM-MCF-7 and MDA-MB-231 cells were transfected with 100 nmol/L siRNA targeting GPR55 for 48 h, whereas untreated control cells were cultured in a 6-well plate and grown to 80% confluence. Then, either 2.5 μmol/L LPI or 2.5 μmol/L CID16020046 was added to the respective culture media. A small area was then disrupted by scratching a line through the monolayer with a needle, and images of the monolayer were captured after the initial scratching and 24 h later to observe cell migration during wound closing. The images were compared to quantify the migration rate of the cells.
Fluorescence staining
Cells were subjected to indirect fluorescence staining as previously described27. Rhodamine phalloidin (Invitrogen, Carlsbad, CA, USA) was used to detect F-actin. Stained cells were observed with a Nikon TE200 inverted microscope, and the resulting images were analyzed using Spot version 4.5 software (Diagnostic Instruments Inc, MI, USA). The number of filopodia per cell and the length of the filopodia in 20 cells in different visual fields were examined by using FiloDetect software28.
Modified Boyden's chamber assay
A modified Boyden's chamber assay was performed27 with slight modifications. LM-MCF-7 and MDA-MB-231 cells were transfected with 50 nmol/L or 100 nmol/L siRNA targeting GPR55 for 48 h, whereas two other groups of cells (104/well) were pretreated with either the GPR55 inhibitor CID16020046 or LPI. All cells were suspended in serum-free RPMI-1640 containing 0.5% bovine serum albumin (RPMI-1640/BSA) and loaded into the upper wells of the chamber (Neuroprobe Inc, Gaithersburg, MD, USA). The lower wells were filled with RPMI-1640/BSA containing 5% FBS. Collagen-coated polycarbonate filters with 8-μm pores were used. After a 10-h incubation, the cells that migrated to the lower surface of the filter were fixed in methanol and stained with Giemsa solution. Each condition was tested in quadruplicate, and the number of cells from five randomly selected high-power fields (HPE; 400× magnification) were counted per well. For the assay, the siRNA, CID16020046 or LPI were added into both the corresponding upper and lower wells at the same concentration. The experiments were repeated 3 times.
Flow cytometry analysis
LM-MCF-7 and MDA-MB-231 cells were transfected with 100 nmol/L siRNA targeting GPR55 for 48 h, whereas two other groups of cells were pretreated with either the 2.5 μmol/L GPR55 inhibitor CID16020046 or 2.5 μmol/L LPI for 24 h. Cells were harvested by trypsinization and washed twice with phosphate buffer saline (PBS). The washed cells were resuspended in 0.6 mL PBS (pH 7.4) and fixed by the addition of 1.4 mL 100% ethanol at 4 °C overnight. Fixed cells were rinsed twice with PBS, re-suspended in propidium iodine solution, which included 50 μg/mL propidium iodide and 50 μg/mL RNaseA (Sigma, MO, USA) in PBS without calcium and magnesium, and then incubated at 37 °C for 30 min in the dark. Stained cells were passed through a nylon-mesh sieve to remove cell clumps and analyzed using a FACScan flow cytometer and CellQuest analysis software (Becton Dickinson, San Jose, CA, USA). The cell proliferative index (PI) was calculated as the sum of S and G2/M phase cells and expressed as a fraction of the total cell population (PI=[(S+G2/M)/(G0/G1+S+G2/M)]×100%). Flow cytometry analysis was repeated three times independently.
Luciferase reporter gene assays
The pGL3-HBXIP and pGL3-Capn4 plasmids and the Renilla luciferase reporter vector pRL-TK were used as previously described22. Cells were seeded onto 24-well plates at a density of 1×104 cells per well and cultured for 24 h at 60%–70% confluence before transfection. The corresponding plasmids were transfected using Lipofectamine™ 2000 according to the manufacturer's instructions (Invitrogen, Carlsbad, CA, USA). The promoter activities of the HBXIP and Capn4 genes were determined 36 h post-transfection using a dual-luciferase reporter gene assay kit (Promega, USA), and promoter luciferase activity was normalized to that of Renilla luciferase. All experiments were performed in triplicate.
Western blot analysis
Western blotting was conducted using standard protocols. MCF-7, LM-MCF-7, and MDA-MB-231 cells were seeded on 6-well plates and made quiescent upon reaching confluence by incubating the cells in serum-free RPMI-1640 containing 2.5 μmol/L LPI for 12 h. Then, either 20 μmol/L ML-7 or 30 μmol/L PD98059 was added to the LM-MCF-7 and MDA-MB-231 cells for 6 h. The cells were washed three times with ice-cold PBS and directly extracted into lysis buffer. The antibodies used in this study targeted HBXIP (Santa Cruz, CA, USA), p-ERK1/2 (phospho-p44/p42 MAPK; Cell Signaling Technology, Danvers, MA, USA), ERK1/2 (p44/p42 MAPK; Cell Signaling Technology, Danvers, MA, USA), JNK (Cell Signaling Technology, Danvers, MA, USA), MLCK (Sigma–Aldrich, USA), p-MLC (pSer19; Sigma–Aldrich), β-catenin (Sigma–Aldrich, USA), and β-actin (Sigma, USA). All the experiments were repeated 3 times.
In vivo metastasis assays
For in vivo metastasis assays, LM-MCF-7 cells transfected with GPR55 siRNA or control siRNA for 48 h were re-suspended in 0.1 mL PBS and injected into the lateral tail vein of 6-week-old Balb/c female mice (4 per group, 5×105 cells per mouse). Two weeks post-injection, the mice were sacrificed, and the lungs were removed and fixed in Bouin's solution for 24 h. The lungs were imaged, and the nodules were counted using a dissecting microscope at 4× magnification. Representative slices were stained with hematoxylin and eosin (H&E). All experiments involving animals complied with protocols approved by the Institutional Animal Care and Use Committee of the Chinese Institute of Laboratory Animal Science (ILAS-GC-2015-002).
Statistical analysis
All data are presented as the mean±standard error of the mean (SEM) and were analyzed using either ANOVA or Student's t-tests with Prism 4.0 (GraphPad Software, CA, USA) software. A P value <0.05 was considered statistically significant. All statistical tests were two sided. The Chi square distribution was used to compare percentages, and correlations between the expression levels of GPR55 and HBXIP in tumor tissues were evaluated using Pearson's correlation coefficient. GPR55 expression levels in primary breast carcinoma and adjacent normal breast tissues were compared using a Wilcoxon signed-rank test.
Results
GPR55 expression is upregulated in metastatic lymph nodes, breast cancer tissues and malignant cancer cells
It has been reported that GPR55 can facilitate cell migration2,29, but the relationship between GPR55 expression and cancer metastasis is not fully understood. We were interested in GPR55 expression levels in breast cancer tissues and metastatic lymph nodes. Therefore, 49 samples of infiltrating primary carcinoma and 38 samples of metastatic lymph nodes were subjected to immunohistochemical staining using tissue arrays from the same tissue paraffin block to detect GPR55 expression. Our data showed that 92.1% (35/38) of the metastatic lymph node tissue samples were positive for GPR55, which was much higher than the percentage of GPR55-positive primary tissue samples (73.4%, 36/49; Chi square test, P=0.0013, Figure 1A), supporting the notion that GPR55 plays an important role in the metastasis of breast cancer.

GPR55 expression is upregulated in clinical metastatic lymph nodes, breast cancer tissues and malignant cancer cells. (A) GPR55 expression was examined by immunohistochemical staining of normal breast tissue, breast cancer tissue and metastatic lymph node tissue. (B) The mRNA levels of GPR55 were examined in clinical breast carcinoma samples and their adjacent normal tissues using qRT-PCR (P=0.0072, Student's t-test, n=36). (C) The mRNA levels of GPR55 were detected in MCF-7, LM-MCF-7 and MDA-MB-231 cells using RT-PCR. (D) GPR55 protein expression was examined in MCF-7, LM-MCF-7 and MDA-MB-231 cells via Western blot analysis. The results are representative of three independent experiments.
Next, using qRT-PCR, we observed that GPR55 mRNA levels were obviously higher in breast tumors than in adjacent normal tissues (n=36) (Figure 1B). Furthermore, we investigated the correlation between GPR55 expression in breast cancer cells and their migratory ability. Interestingly, GPR55 mRNA and protein expression levels were significantly upregulated in LM-MCF-7 (a high metastasis subclone of MCF-7 cell line27) and MDA-MB-231 cells compared with MCF-7 cells (Figure 1C and 1D). Our data indicated that the expression levels of GPR55 were significantly and positively correlated with the migratory ability of breast cancer cells.
LPI content in breast cancer patients' plasma
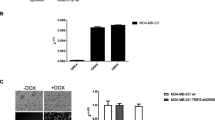
LPI has been identified as an endogenous GPR55 ligand1, as it activates GPR55 in cells. To clarify the role of GPR55 in breast cancer progression and metastasis, endogenous LPI levels in the plasma of 36 breast cancer patients and 14 healthy individuals were examined using LC-MS. The mass spectrometry data revealed that LPI (18:0) levels were significantly increased in the plasma samples of all 36 breast cancer patients compared to the samples of healthy individuals (P=0.0039, Student's t-test, Figure 2A). Total LPI (16:0, 18:0, 24:0) was also increased in the plasma of breast cancer patients, but this result was not as significant (P=0.047, Student's t-test, Figure 2B). Our results support the notion that LPI and its receptor GPR55 are associated with breast cancer carcinogenesis.

LPI content in the plasma of breast cancer patients. LPI, an endogenous ligand of GPR55, was evaluated in the blood samples of breast cancer patients (n=36) and healthy subjects (control; n=14) using LC-MS, (A) LPI (18:0) was significantly increased in breast cancer patients compared with healthy individuals (**P=0.0039, Student's t-test). (B) Total LPI (16:0, 18:0, 24:0) was slightly increased compared with controls (*P=0.047, Student's t test).
GPR55 is involved in the migration of breast cancer cells and filopodia formation
To identify the role of the GPR55/LPI axis in the migration of breast cancer cells, we investigated cell motility based on a wound healing assay and the modified Boyden's chamber assay. The results of the wound healing assay revealed that 2.5 μmol/L LPI increased the migratory ability of LM-MCF-7 and MDA-MB-231 cells (Figure 3B). Similarly, the modified Boyden's chamber assay showed that LM-MCF-7 and MDA-MB-231 cells treated with 2.5 μmol/L LPI exhibited 3.51-fold (P=0.0071, Student's t-test) and 3.73-fold (P=0.0055, Student's t-test) increased motility, respectively, compared to control cells (Figure 3C and 3D). Incubating LM-MCF-7 and MDA-MB-231 cells with 2.5 μmol/L CID16020046 (a GPR55-specific inhibitor) in the wound healing assay or with either 1 μmol/L or 2.5 μmol/L CID16020046 in the modified Boyden's chamber assay significantly reduced the migratory abilities in a dose-dependent manner (Figure 3B, 3C and 3D). Likewise, GPR55 knockdown via transfection with 50 nmol/L or 100 nmol/L siRNA targeting GPR55 (Figure 2A) significantly decreased LPI-induced LM-MCF-7 and MDA-MB-231 cell migration in a dose-dependent manner (Figure 3B, 3C and 3D). Therefore, our results indicated that the GPR55/LPI axis exerts a direct effect on controlling the migration of breast cancer cells.

GPR55 is involved in breast cancer cell migration and increased filopodia formation. (A) The GPR55 protein levels in LM-MCF-7 and MDA-MB-231 cells transfected with siGPR55 were examined via Western blot analysis. (B) The migratory ability of LM-MCF-7 and MDA-MB-231 cells treated with 2.5 μmol/L LPI, 2.5 μmol/L CID16020046 or siGPR55 was examined based on a wound healing assay. The results were expressed as relative wound width (**P<0.01 vs control, Student's t-test). (C) The migratory ability of LM-MCF-7 cells with different treatments was determined using the modified Boyden's chamber assay (columns reflect the mean of six fields in which at least 100 cells were counted; *P<0.05, **P<0.01 vs control, Student's t-test). The results are representative of three independent experiments.

(D) The migratory ability of MDA-MB-231 cells with different treatments was determined using the modified Boyden's chamber assay (columns reflect the mean of six fields in which at least 100 cells were counted; *P<0.05, **P<0.01 vs control, Student's t-test). The results are representative of three independent experiments. (E) F-actin was visualized using immunofluorescence staining in cells treated with 2.5 μmol/L LPI, 2.5 μmol/L CID16020046 or siGPR55. Filopodia protrusions are indicated by the arrows. Scale bar, 10 μm.
Filopodia are a fundamental characteristic of cancer cell migration30. Examining F-actin can be used to evaluate filopodia, and the number and the length of filopodia can be quantified using FiloDetect software by monitoring the changes in filopodia formation in the cells31. Fluorescence staining showed that LPI-induced filopodia formation could be disrupted in LM-MCF-7 and MDA-MB-231 cells by treatment with either siRNA targeting GPR55 or 2.5 μmol/L CID16020046 (Figure 2E). Thus, we concluded that the GPR55/LPI axis can increase filopodia formation to promote breast cancer cell migration.
HBXIP expression levels are significantly and positively associated with GPR55 levels in breast cancer
It has been reported that HBXIP can facilitate the migration of cancer cells32. Thus, we sought to determine the relationship between GPR55 and HBXIP expression during cancer cell migration. The luciferase reporter gene assay showed that blocking GPR55 activity with CID16020046 inhibited the transcriptional activity of the HBXIP promoter in both LM-MCF-7 cells and MDA-MB-231 cells in a dose-dependent manner (Figure 4A). Likewise, HBXIP promoter activity was inhibited by knocking down GPR55 via transfection of breast cancer cells with GPR55-targeted siRNA in a dose-dependent manner (Figure 4B). These results support the idea that HBXIP serves as an important downstream factor of GPR55 in regulating breast cancer cell migration. Moreover, we investigated the correlation between GPR55 and HBXIP expression in breast tumor tissues. Our data showed that GPR55 expression levels exhibited a significant and positive correlation with HBXIP expression levels in breast cancer tissues (Wilcoxon signed-rank test, Figure 4C). The correlation between HBXIP and GPR55 in 8 breast cancer cell lines was further confirmed according to the Spearman correlation coefficient of 0.9347 (P<0.01, Figure 4D). Taken together, these data led us to conclude that HBXIP expression levels are significantly and positively associated with GPR55 expression levels in breast cancer tissues.

HBXIP expression is significantly and positively associated with GPR55 expression in breast cancer. (A) HBXIP promoter activity was examined using dual-luciferase reporter gene assays in LM-MCF-7 and MDA-MB-231 cells treated with CID16020046 (*P<0.05, **P<0.01 vs control, Student's t-test). (B) HBXIP promoter activity was analyzed using dual-luciferase reporter gene assays in LM-MCF-7 and MDA-MB-231 cells treated with siRNA targeting GPR55. (C) The correlation between GPR55 and HBXIP expression was analyzed using qRT-PCR in the abovementioned tumor tissues (n=36, P<0.01, Wilcoxon signed-rank test). (D) The correlation between HBXIP mRNA levels and GPR55 mRNA levels was assessed in 8 human breast cancer cell lines (R2=0.9347, P<0.001).
The LPI/GPR55 axis promotes breast cancer cell migration by activating HBXIP/p-ERK1/2/Capn4 and MLCK/p-MLC signaling
We recently reported that factors such as HBXIP, p-ERK1/2, p-JNK, MLCK, p-MCL, β-catenin and Capn4 are responsible for the enhanced migratory abilities of LM-MCF-7 and MDA-MB-231 cells compared with MCF-7 cells27,32,33. Therefore, we sought to determine whether any of these factors participate in LPI/GPR55 axis-induced migration of breast cancer cells via GPR55. We examined the expression of HBXIP, p-ERK1/2, ERK1/2, p-JNK, MLCK, p-MLC, β-catenin, and Capn4 after incubating cells with 2.5 μmol/L LPI. The results showed that HBXIP, p-ERK1/2, MLCK, p-MLC and Capn4 were upregulated relative to control cells but that the expression levels of p-JNK and β-catenin were not affected by LPI (Figure 5A). Next, either treatment with CID16020046 or knocking down GPR55 via targeted siRNA transfection led to downregulation of HBXIP, MLCK, p-MLC, p-ERK1/2 and Capn4 in a dose-dependent manner but failed to affect the levels of p-JNK and β-catenin (Figure 5A). Similar results were verified in MDA-MB-231 cells (Figure 5B), suggesting that elevated expression of these effectors may play an important role in LPI/GPR55-mediated signaling.

The LPI/GPR55 axis promotes breast cancer cell migration by activating HBXIP/p-ERK1/2/Capn4 and MLCK/p-MLC signaling. Examination of the expression levels of p-ERK1/2, ERK1/2, HBXIP, p-JNK, MLCK, p-MLC, β-catenin and Capn4 in LM-MCF-7 cells (A) and MDA-MB-231 cells (B) treated with various doses of CID16020046 and siGPR55. (C) HBXIP silencing reduced the expression of p-ERK1/2 and Capn4 in both LM-MCF-7 and MDA-MB-231 cells treated with 2.5 μmol/L LPI. (D) The expression levels of HBXIP, p-ERK1/2 and Capn4 were not influenced by the dephosphorylation of MLC via treating cells with 20 μmol/L ML-7.
Because HBXIP expression is significantly and positively associated with GPR55 expression in breast cancer, we hypothesized that the LPI/GPR55 axis regulates MLCK, p-MLC, p-ERK1/2 and Capn4 in breast cancer cells by regulating HBXIP. Our findings showed that silencing HBXIP reduced p-ERK1/2 and Capn4 expression in both LM-MCF-7 and MDA-MB-231 cells pretreated with LPI but did not affect MLCK and p-MLC expression (Figure 5C). Interestingly, blocking MLC phosphorylation using 20 μmol/L ML-733 had no influence on the expression levels of HBXIP, p-ERK1/2 or Capn4 (Figure 5D) and reducing p-ERK1/2 levels with 30 μmol/L PD9805927 led to Capn4 downregulation (Figure 5E). Moreover, silencing HBXIP reduced Capn4 promoter activity in both malignant breast cancer cell lines pretreated with 2.5 μmol/L LPI (Figure 5F) in a dose-dependent manner. Taken together, these data suggest that the GPR55/LPI axis mediates breast cancer cell migration via HBXIP/p-ERK1/2/Capn4 and MLCK/p-MLC signaling, which are mutually exclusive pathways.

(E) Downregulation of p-ERK1/2 by PD98059 reduced the expression of Capn4 in both LM-MCF-7 and MDA-MB-231 cells pretreated with 2.5 μmol/L LPI. The results are representative of three independent experiments. (F) HBXIP silencing dose-dependently reduced the promoter activities of Capn4 in both malignant breast cancer cell lines.
GPR55 expression affects breast cancer cell lung metastasis in animal models
The profound effects of GPR55 on breast cancer cell migration in vitro led us to examine its influence on breast cancer metastasis in vivo. LM-MCF-7 cells, a metastatic subclone from the MCF-7 breast cancer cell line (obtained Chinese patent: 031302645), form tumors in the lungs and other organs after tail vein injection21. LM-MCF-7 cells transfected with GPR55 siRNA or control siRNA were intravenously injected into the lateral tail vein of 6-week-old Balb/c female mice. The mice were sacrificed 2 weeks later, the lungs were removed, and the number of metastatic foci on the lungs was assessed. As shown in Figure 6A and 6B, control LM-MCF-7 cells produced large and numerous metastatic foci. Downregulating GPR55 by RNAi markedly reduced the number of lung metastases (P<0.01 vs control). Representative histological photomicrographs of lung sections stained with H&E are shown in Figure 6C. This profound reduction in the number of metastatic lesions suggests that loss of GPR55 mainly affects breast cancer cell metastasis and the formation of metastatic foci. However, GPR55 could also slightly affect metastatic outgrowth as it affects the proliferation of breast cancer cells (Supplementary Figure S1).

Inhibition of GPR55 in LM-MCF-7 cells substantially inhibited lung metastasis. (A) C57/BL6 mice were intravenously injected with 5×105 transfected cells, and pulmonary metastasis was evaluated after 14 d. Representative photographs of lungs from each group are shown. (B) Quantitative evaluation of detectable nodules on the entire lungs. Data are expressed as the mean±SD (**P<0.05). (C) Representative histological photomicrographs of lung sections stained with H&E.
Discussion
GPCRs activated by lysophospholipid ligands, such as LPA receptors, are regarded as important players in cancer development34. This raises the possibility that other lysophospholipid ligand-responsive GPCRs such as GPR55 could exert tumorigenic behavior in a similar manner. Parallel to this concept, high levels of the endogenous ligands of these GPCRs may contribute to carcinogenesis. For example, LPI, an endogenous GPR55 ligand, has been reported to be elevated in the blood of patients with ovarian cancer13; therefore, the pathophysiological function of the GPR55/LPI axis has gained increasing attention as a potential driving force of cancer. In the present study, we demonstrated that the GPR55/LPI axis plays an important role in the migratory behavior of breast cancer cells.
To explore the role of the LPI/GPR55 axis in breast cancer cells, we examined GPR55 expression in metastatic lymph nodes from patients with breast cancer. Notably, we found that these metastatic lymph nodes exhibited a high frequency of positive GPR55 expression. We detected significantly increased LPI (18:0) content in plasma samples from breast cancer patients. Therefore, activation of the LPI/GPR55 axis is highly likely in breast cancer patients who present increased levels of plasma LPI, as observed in our study. A previous study showed that stearic (18:0) acid constitutes one of the major fatty acid components of LPI in the serum of healthy individuals and patients with ovarian cancer13, which may explain why we observed significant differences in the LPI (18:0) species. Various lines of evidence point to a role of endogenous LPI in cancer. Clinical data implicate that malignant ascites from malignant ovarian tissue samples contain higher levels of LPI than ascites collected from non-malignant samples12. Regarding cancer, recent data suggest that GPR55 may be part of the molecular circuitry controlling tumor growth. Thus, this receptor promotes glioma, breast1, prostate and ovarian3 cancer cell proliferation as well as breast cancer cell migration2 in culture. GPR55 also induced tumor growth in a xenograft-based model of glioma1. Our data are consistent with these reports, and additionally, we found that GPR55 expression levels were significantly and positively associated with the migratory ability of breast cancer cells and that LPI significantly promoted breast cancer cell migration. Moreover, siRNA knockdown of GPR55 clearly ameliorated CID16020046-induced inhibition of migration, as assessed using chemotactic assays, indicating a major role of GPR55 in the LPI-mediated migratory effects. Based on the results of the mouse xenograft model, the profound reduction in the number of metastatic lesions suggests that loss of GPR55 mainly affects breast cancer cell metastasis and the formation of metastatic foci (Figure 6). These data clearly indicate a crucial role of the LPI/GPR55 axis in breast cancer cell migration. Our data are in agreement with previous reports that treating HCT116 cells (a colon cancer cell line) with LPI enhanced the migratory ability of the cells35. Activation of the LPI/GPR55 axis likely occurs in breast cancer patients who exhibit increased levels of plasma LPI, as observed in our study. High levels of LPI can easily influence the interaction of cancer cells with the endothelium because LPI is not only produced in endothelial and cancer cells but also in neutrophils and platelets, ie, cells that assist invading tumor cells35.
We next attempted to identify the mechanism by which the LPI/GPR55 axis promotes breast cancer cell migration. In this study, we showed that the LPI/GPR55 axis upregulated HBXIP at the promoter and protein levels in breast cancer cells. Moreover, HBXIP expression was significantly and positively associated with GPR55 expression in breast cancer tissues. We previously reported that the oncoprotein HBXIP acts as an important regulator of breast cancer cell migration. HBXIP can regulate IL-8 transcription via NF-κB activation; overexpression of HBXIP promotes the migration of MCF-7 cells36, and miR-520b suppresses MDA-MB-231 and LM-MCF-7 migration by targeting HBXIP36. Moreover, HBXIP upregulates S100A4 by activating the S100A4 promoter via STAT4 and inducing PTEN/PI3K/AKT signaling to promote the growth and migration of breast cancer cells20. Cell migration requires sustained forward progression of the plasma membrane at the front of the cell, and these membrane protrusions (ie, lamellipodia and filopodia) directly push the plasma membrane and move the cells37. HBXIP can enhance breast cancer cell migration by increasing filopodia formation via MEKK2/ERK1/2/Capn4 signaling32. According to the abovementioned reports and our present results, we revealed that the GPR55/LPI axis induces filopodia formation and enhances breast cancer cell migration by upregulating the oncoprotein HBXIP.
It has been reported that the reciprocal cross-talk of MLCK and the activated ERK1/2 signaling pathway promote breast cancer cell migration by which both phosphorylated myosin light chain (p-MLC) and β-catenin serve as essential downstream effectors27. Based on the results of assays involving siRNA and inhibitor treatments, our findings suggest that the GPR55/LPI axis mediates breast cancer cell migration via the HBXIP/p-ERK1/2/Capn4 and MLCK/p-MLC pathways, two mutually exclusive signaling pathways. Previous studies showed that p-ERK1/2 plays an important role in regulating a wide array of cellular functions, including cell motility27. Many proteins, such as Rho, Rac1, and Cdc42, are involved in regulating cell migration through the ERK1/2 signaling pathway38. Furthermore, Capn4 plays a key role in tumor migration39 and can regulate cell-substrate mechanical interactions during fibroblast migration40. p-ERK1/2 also activates Capn4 to promote the proliferation of breast cancer cells41. Moreover, we reported that Capn4 promotes the migration of hepatocellular carcinoma by activating NF-κB signaling42. In this study, we revealed that upregulating p-ERK1/2 and Capn4 might control GPR55-enhanced HBXIP expression. Phosphorylation of MLC20 is essential for triggering actin–myosin interactions43. Many agents that induce MLC20 phosphorylation can cause cell migration44. In the present study, we found that the GPR55/LPI axis can potently upregulate MLCK expression and promote MLC phosphorylation. Thus, our results revealed that the GPR55/LPI axis promotes breast cancer cell migration through the HBXIP/ERK1/2/Capn4 and MLCK/p-MLC signaling pathways.
Taken together, our data demonstrate that GPR55 is involved in the migratory behavior of breast cancer cells and that this activity is impeded by blocking GPR55 signaling. Considering the increase in LPI levels in breast cancer patients and the presence of GPR55 in breast tumor cells, tumor tissues and metastatic lymph nodes, the LPI/GPR55 axis could contribute to breast cancer cell metastasis. Furthermore, the LPI/GPR55 axis promotes cell migration via two mutually exclusive pathways: the p-ERK1/2/Capn4 and MLCK/MLC pathways. Our findings provide new insight into the mechanism of breast cancer cell migration. Thus, GPR55 may serve as a novel therapeutic target for treating breast cancer.
Author contribution
Xiao-lei ZHOU conceived and designed the study; Yu-pin SONG, Xin GUO, and Chong-yue ZHU performed the experiments; Xiao-lei ZHOU wrote the paper; Wei ZOU reviewed and edited the manuscript. All authors read and approved the manuscript.
Abbreviations
LPI, L-α-lysophosphatidylinositol; GPR55, G protein coupled receptor 55; CID16020046, 4-[4-(3-hydroxyphenyl)-3-(4-methylphenyl)-6-oxo-1H,4H,5H,6H-pyrrolo [3,4-c] pyrazol-5-yl] benzoic acid; HBXIP, hepatitis B X-interacting protein; Capn4, Calpain small subunit 1; ERK1/2, extracellular signal-regulated kinase 1/2.
References
Andradas C, Caffarel MM, Perez-Gomez E, Salazar M, Lorente M, Velasco G, et al. The orphan G protein-coupled receptor GPR55 promotes cancer cell proliferation via ERK. Oncogene 2011; 30: 245–52.
Ford LA, Roelofs AJ, Anavi-Goffer S, Mowat L, Simpson DG, Irving AJ, et al. A role for L-alpha-lysophosphatidylinositol and GPR55 in the modulation of migration, orientation and polarization of human breast cancer cells. Br J Pharmacol 2010; 160: 762–71.
Pineiro R, Maffucci T, Falasca M . The putative cannabinoid receptor GPR55 defines a novel autocrine loop in cancer cell proliferation. Oncogene 2011; 30: 142–52.
Perez-Gomez E, Andradas C, Flores JM, Quintanilla M, Paramio JM, Guzman M, et al. The orphan receptor GPR55 drives skin carcinogenesis and is upregulated in human squamous cell carcinomas. Oncogene 2013; 32: 2534–42.
Henstridge CM, Balenga NA, Kargl J, Andradas C, Brown AJ, Irving A, et al. Minireview: recent developments in the physiology and pathology of the lysophosphatidylinositol-sensitive receptor GPR55. Mol Endocrinol 2011; 25: 1835–48.
Ross RA . L-alpha-lysophosphatidylinositol meets GPR55: a deadly relationship. Trends Pharmacol Sci 2011; 32: 265–9.
Oka S, Nakajima K, Yamashita A, Kishimoto S, Sugiura T . Identification of GPR55 as a lysophosphatidylinositol receptor. Biochem Biophys Res Commun 2007; 362: 928–34.
Lauckner JE, Jensen JB, Chen HY, Lu HC, Hille B, Mackie K . GPR55 is a cannabinoid receptor that increases intracellular calcium and inhibits M current. Proc Natl Acad Sci U S A 2008; 105: 2699–704.
Whyte LS, Ryberg E, Sims NA, Ridge SA, Mackie K, Greasley PJ, et al. The putative cannabinoid receptor GPR55 affects osteoclast function in vitro and bone mass in vivo. Proc Natl Acad Sci U S A 2009; 106: 16511–6.
Oka S, Kimura S, Toshida T, Ota R, Yamashita A, Sugiura T . Lysophosphatidylinositol induces rapid phosphorylation of p38 mitogen-activated protein kinase and activating transcription factor 2 in HEK293 cells expressing GPR55 and IM-9 lymphoblastoid cells. J Biochem 2010; 147: 671–8.
Moreno-Navarrete JM, Catalan V, Whyte L, Diaz-Arteaga A, Vazquez-Martinez R, Rotellar F, et al. The L-alpha-lysophosphatidylinositol/GPR55 system and its potential role in human obesity. Diabetes 2012; 61: 281–91.
Xiao Y, Chen Y, Kennedy AW, Belinson J, Xu Y . Evaluation of plasma lysophospholipids for diagnostic significance using electrospray ionization mass spectrometry (ESI-MS) analyses. Ann N Y Acad Sci 2000; 905: 242–59.
Shen Z, Wu M, Elson P, Kennedy AW, Belinson J, Casey G, et al. Fatty acid composition of lysophosphatidic acid and lysophosphatidylinositol in plasma from patients with ovarian cancer and other gynecological diseases. Gynecol Oncol 2001; 83: 25–30.
Falasca M, Silletta MG, Carvelli A, Di Francesco AL, Fusco A, Ramakrishna V, et al. Signalling pathways involved in the mitogenic action of lysophosphatidylinositol. Oncogene 1995; 10: 2113–24.
Marusawa H, Matsuzawa S, Welsh K, Zou H, Armstrong R, Tamm I, et al. HBXIP functions as a cofactor of survivin in apoptosis suppression. EMBO J 2003; 22: 2729–40.
Wen Y, Golubkov VS, Strongin AY, Jiang W, Reed JC . Interaction of hepatitis B viral oncoprotein with cellular target HBXIP dysregulates centrosome dynamics and mitotic spindle formation. J Biol Chem 2008; 283: 2793–803.
Liu Q, Bai X, Li H, Zhang Y, Zhao Y, Zhang X, et al. The oncoprotein HBXIP upregulates Lin28B via activating TF II D to promote proliferation of breast cancer cells. Int J Cancer 2013; 133: 1310–22.
Xu F, You X, Liu F, Shen X, Yao Y, Ye L, et al. The oncoprotein HBXIP up-regulates Skp2 via activating transcription factor E2F1 to promote proliferation of breast cancer cells. Cancer Lett 2013; 333: 124–32.
Yue L, Li L, Liu F, Hu N, Zhang W, Bai X, et al. The oncoprotein HBXIP activates transcriptional coregulatory protein LMO4 via Sp1 to promote proliferation of breast cancer cells. Carcinogenesis 2013; 34: 927–35.
Liu S, Li L, Zhang Y, Zhang Y, Zhao Y, You X, et al. The oncoprotein HBXIP uses two pathways to up-regulate S100A4 in promotion of growth and migration of breast cancer cells. J Biol Chem 2012; 287: 30228–39.
You J, Mi D, Zhou X, Qiao L, Zhang H, Zhang X, et al. A positive feedback between activated extracellularly regulated kinase and cyclooxygenase/lipoxygenase maintains proliferation and migration of breast cancer cells. Endocrinology 2009; 150: 1607–17.
Zhang F, Wang Q, Ye L, Feng Y, Zhang X . Hepatitis B virus X protein upregulates expression of calpain small subunit 1 via nuclear factor-kappaB/p65 in hepatoma cells. J Med Virol 2010; 82: 920–8.
Kong GY, Zhang JP, Zhang S, Shan CL, Ye LH, Zhang XD . Hepatitis B virus X protein promotes hepatoma cell proliferation via upregulation of MEKK2. Acta Pharmacol Sin 2011; 32: 1173–80.
Matyash V, Liebisch G, Kurzchalia TV, Shevchenko A, Schwudke D . Lipid extraction by methyl-tert-butyl ether for high-throughput lipidomics. J Lipid Res 2008; 49: 1137–46.
Fauland A, Kofeler H, Trotzmuller M, Knopf A, Hartler J, Eberl A, et al. A comprehensive method for lipid profiling by liquid chromatographyion cyclotron resonance mass spectrometry. J Lipid Res 2011; 52: 2314–22.
Wang Y, Cui M, Cai X, Sun B, Liu F, Zhang X, et al. The oncoprotein HBXIP up-regulates SCG3 through modulating E2F1 and miR-509-3p in hepatoma cells. Cancer Lett 2014; 352: 169–78.
Zhou X, Liu Y, You J, Zhang H, Zhang X, Ye L . Myosin light-chain kinase contributes to the proliferation and migration of breast cancer cells through cross-talk with activated ERK1/2. Cancer Lett 2008; 270: 312–27.
Nilufar S, Morrow AA, Lee JM, Perkins TJ . FiloDetect: automatic detection of filopodia from fluorescence microscopy images. BMC Syst Biol 2013; 7: 66.
Paul RK, Wnorowski A, Gonzalez-Mariscal I, Nayak SK, Pajak K, Moaddel R, et al. (R,R')-4'-methoxy-1-naphthylfenoterol targets GPR55-mediated ligand internalization and impairs cancer cell motility. Biochem Pharmacol 2014; 87: 547–61.
Blanchoin L, Boujemaa-Paterski R, Sykes C, Plastino J . Actin dynamics, architecture, and mechanics in cell motility. Physiol Rev 2014; 94: 235–63.
Xue F, Janzen DM, Knecht DA . Contribution of filopodia to cell migration: A mechanical link between protrusion and contraction. Int J Cell Biol 2010; 2010: 507821.
Li Y, Zhang Z, Zhou X, Li L, Liu Q, Wang Z, et al. The oncoprotein HBXIP enhances migration of breast cancer cells through increasing filopodia formation involving MEKK2/ERK1/2/Capn4 signaling. Cancer Lett 2014; 355: 288–96.
Zhou XL, Qin XR, Zhang XD, Ye LH . Downregulation of Dickkopf-1 is responsible for high proliferation of breast cancer cells via losing control of Wnt/beta-catenin signaling. Acta Pharmacol Sin 2010; 31: 202–10.
Dorsam RT, Gutkind JS . G-protein-coupled receptors and cancer. Nat Rev Cancer 2007; 7: 79–94.
Kargl J, Andersen L, Hasenohrl C, Feuersinger D, Stancic A, Fauland A, et al. GPR55 promotes migration and adhesion of colon cancer cells indicating a role in metastasis. Br J Pharmacol 2016; 173: 142–54.
Hu N, Zhang J, Cui W, Kong G, Zhang S, Yue L, et al. miR-520b regulates migration of breast cancer cells by targeting hepatitis B X-interacting protein and interleukin-8. J Biol Chem 2011; 286: 13714–22.
Ridley AJ . Life at the leading edge. Cell 2011; 145: 1012–22.
Kamai T, Yamanishi T, Shirataki H, Takagi K, Asami H, Ito Y, et al. Overexpression of RhoA, Rac1, and Cdc42 GTPases is associated with progression in testicular cancer. Clin Cancer Res 2004; 10: 4799–805.
Zheng PC, Chen X, Zhu HW, Zheng W, Mao LH, Lin C, et al. Capn4 is a marker of poor clinical outcomes and promotes nasopharyngeal carcinoma metastasis via nuclear factor-kappaB-induced matrix metalloproteinase 2 expression. Cancer Sci 2014; 105: 630–8.
Undyala VV, Dembo M, Cembrola K, Perrin BJ, Huttenlocher A, Elce JS, et al. The calpain small subunit regulates cell-substrate mechanical interactions during fibroblast migration. J Cell Sci 2008; 121: 3581–8.
Wang GS, Huang YG, Li H, Bi SJ, Zhao JL . ERK/CANP rapid signaling mediates 17beta-estradiol-induced proliferation of human breast cancer cell line MCF-7 cells. Int J Clin Exp Med 2014; 7: 156–62.
Zhang X, You X, Wang Q, Zhang T, Du Y, Lv N, et al. Hepatitis B virus X protein drives multiple cross-talk cascade loops involving NF-kappaB, 5-LOX, OPN and Capn4 to promote cell migration. PLoS One 2012; 7: e31458.
Lai JM, Hsieh CL, Chang ZF . Caspase activation during phorbol ester-induced apoptosis requires ROCK-dependent myosin-mediated contraction. J Cell Sci 2003; 116: 3491–501.
Gutjahr MC, Rossy J, Niggli V . Role of Rho, Rac, and Rho-kinase in phosphorylation of myosin light chain, development of polarity, and spontaneous migration of Walker 256 carcinosarcoma cells. Exp Cell Res 2005; 308: 422–38.
Acknowledgements
This project was supported by grants from the National Natural Science Foundation of China (No 81602428 and 81402490) and the Colleges and Universities in Hebei Province Science and Technology Research Project (No QN2016020), and Science and Technology Research and Developmental Guidance Program of Shijiazhuang City (No 171201073A).
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary files are available at the website of Acta Pharmacologica Sinica.
Supplementary information
Supplementary information
Supplementary Table and Figure (DOC 83 kb)
Rights and permissions
About this article

Cite this article
Zhou, Xl., Guo, X., Song, Yp. et al. The LPI/GPR55 axis enhances human breast cancer cell migration via HBXIP and p-MLC signaling. Acta Pharmacol Sin 39, 459–471 (2018). https://doi.org/10.1038/aps.2017.157
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2017.157
Keywords
This article is cited by
-
The lipidomic profile of the tumoral periprostatic adipose tissue reveals alterations in tumor cell’s metabolic crosstalk
BMC Medicine (2022)
-
The oncoprotein HBXIP competitively binds KEAP1 to activate NRF2 and enhance breast cancer cell growth and metastasis
Oncogene (2019)
-
The oncoprotein HBXIP promotes human breast cancer growth through down-regulating p53 via miR-18b/MDM2 and pAKT/MDM2 pathways
Acta Pharmacologica Sinica (2018)
