
Abstract
Diterpene ginkgolides meglumine injection (DGMI) is a therapeutic extract of Ginkgo biloba L, which has been used for the treatment of cerebral ischemic stroke in China. Ginkgolides A, B and C are the main components of DGMI. This study was designed to investigate the neuroprotective effects of DGMI components against ischemic stroke in vivo and in vitro. Acute cerebral ischemic injury was induced in rats by occlusion of the middle cerebral artery (MCA) for 1.5 h followed by 24 h reperfusion. The rats were treated with DGMI (1, 3 and 10 mg/kg, iv) at the onset of reperfusion and 12 h after reperfusion. Administration of DGMI significantly decreased rat neurological deficit scores, reduced brain infarct volume, and induced protein kinase B (Akt) phosphorylation, which prompted the nuclear translocation of nuclear factor-erythroid 2-related factor 2 (Nrf2) and phosphorylation of the survival regulatory protein cyclic AMP-responsive element binding protein (CREB). Nrf2 activation led to expression of the downstream protein heme oxygenase-1 (HO-1). In addition, PC12 cells were subjected to oxygen-glucose deprivation/reperfusion (OGD/R) in vitro, treatment with DGMI (1, 10 and 20 μg/mL) or ginkgolides A, B or C (10 μmol/L for each) significantly reduced PC12 cell death and increased phosphorylation of Akt, nuclear translocation of Nrf2 and activation of CREB. Activation of Nrf2 and CREB could be reversed by co-treatment with a phosphoinositide-3-kinase (PI3K) inhibitor LY294002. These observations suggest that ginkgolides act as novel extrinsic regulators activating both Akt/Nrf2 and Akt/CREB signaling pathways, protecting against cerebral ischemia/reperfusion (I/R) damage in vivo and in vitro.
Similar content being viewed by others


Introduction
Ischemic stroke is one of the leading causes of death and severe disability around the world; however, effective pharmacological treatment for this condition is lacking1. Diterpene ginkgolides (ginkgolides A, B and C, Figure 1) are the main components of the diterpene ginkgolides meglumine injection (DGMI), extracted from the traditional Chinese medicine Ginkgo biloba L, and have been used clinically for the treatment of cerebral ischemic stroke in China.

Chemical structures of Ginkgolide A, B and C. A, Ginkgolide A. B, Ginkgolide B. C, Ginkgolide C.
The PI3K/Akt signaling pathway has been reported to act as a key regulator in cell death and survival. In ischemic stroke, phosphorylation of Akt leads to further phosphorylation of various downstream proteins, including Nrf2 and CREB, that protect cells from ischemic damage. CREB, as a nuclear transcription factor, is activated by Akt through phosphorylation of Ser1332,3,4,5, facilitating CREB-binding protein (CBP) translocation to the promoter and triggering expression of protective proteins. Among them, the apoptosis regulator Bcl-26,7,8 prevents activation of apoptosis by inhibiting the release of cytochrome c, production of reactive oxygen species (ROS) and the loss of mitochondrial membrane potential (MMP). Nrf2 regulates the transcription and expression of various antioxidant proteins through interaction with the antioxidant response element (ARE), one of the most important reported endogenous antioxidant stress pathways9. As a member of the Cap'n'Collar (CNC) transcription factor family, Nrf2 has a highly conserved basic leucine zipper (bZip) structure. Normally, Nrf2 binds to the Kelch-like ECH-associated protein-1 (Keap1) in the cytoplasm and is degraded through the Cullin 3-based E3-ligase ubiquitination complex-mediated pathway. Once stimulated by external stimuli or ROS, Nrf2 is released from the complex and translocates to the nucleus, where it binds musculoaponeurotic fibrosarcoma (Maf) protein to form a heterodimer and then communicates with ARE to activate the transcription of related phase II detoxifying antioxidant enzyme genes10,11,12. HO-1 is one of the phase II detoxifying enzymes and plays important roles in anti-oxidation protection in ischemia/reperfusion13,14. Upregulation of HO-1 by drug treatment has shown neuroprotective effects both in vivo and in vitro 15,16. These findings indicate that the Nrf2 antioxidant response pathway protects the brain against I/R injury17. Therefore, regulation of Akt/Nrf2 and Akt/CREB signaling pathways might be a promising strategy for the treatment of ischemic stroke.
Ginkgo biloba L, also known as Ginkgo, has various medical effects as a traditional Chinese medicine. It was recorded in the “Compendium of Materia Medica” that Ginkgo had the effect of “dispersing toxin”18. The ancient Chinese phrase “dispersing toxin” now means clearance of inflammation and oxidative state in the human body. The phrase “dispersing toxin” led to the original ideas for today's anti-inflammation and anti-oxidation studies of Ginkgo in stroke. The “Encyclopedia of Chinese Herbal Medicines” is an assembly of Chinese herbal medicines used in ancient China. In this book, the nature product Ginkgo was recorded for the treatment of brain stroke and myocardial infarction19. Currently, Ginkgo biloba L. extracts have shown various pharmacological effects, especially protective effects against I/R injury20,21,22. DGMI and EGb761 are two clinically used therapeutic extracts of Ginkgo biloba L. in China. Ginkgolides were first isolated by Nakanishia23 and have shown various medical effects, including therapeutic action on myocardial injury and stroke24,25. It has been reported that ginkgolide B, one of the main constituents of DGMI, decreases cisplatin-induced ROS generation by reducing NADPH oxidase 2 (NOX2) expression and inducing Nrf2 translocation26. Therefore, in this study, the protective effects of DGMI and its single components (ginkgolides A, B and C) against I/R were investigated both in vitro and in vivo. Underlying molecular mechanisms were also investigated.
Materials and methods
Reagents and chemicals
DGMI was obtained from Jiangsu Kanion Pharmaceutical Co Ltd (Lianyungang, China) with national medicine permission number Z20120024 and batch number 140608, containing ginkgolide A 35%, ginkgolide B 60%, ginkgolide C 2%, ginkgolide K 2%, and 5 mg total ginkgolides per mL, analyzed by an HPLC-ELSD method that we previously developed27. Ginkgolides A, B and C (purity≥98%) were purchased from Shanghai Yuanye Bio-Technology Co Ltd (Shanghai, China). A 10 mmol/L stock solution of ginkgolides A, B and C was prepared in dimethyl sulfoxide (DMSO) and stored at −80 °C. Edaravone injection was purchased from Simcere Pharmaceutical Company (Nanjing, China) with national medicine permission number H20031341 and batch number 040285. The suture used in the middle cerebral artery occlusion/reperfusion (MCAO/R) model was purchased from Cinontech Co Ltd (Beijing, China). The TUNEL apoptosis detection kit was purchased from Roche (Mannheim, Germany). Hoechst 33258, DAPI and 3-[4,5-dimethyl-2-thiazolyl]-2,5-diphenyl-2H-tetrazolium bromide (MTT) were purchased from Sigma-Aldrich (MO, USA). Antibodies against cleaved caspase 3, Bcl-2, Bax, Akt, p-Akt and GAPDH were purchased from Cell Signaling Technology (MA, USA). Antibodies against Nrf2, CREB, p-CREB, HO-1 and histone H3 were purchased from Santa Cruz Biotechnology (CA, USA). LY294002 (a PI3K inhibitor) was purchased from Beyotime Institute of Biotechnology (Beijing, China). Other reagents and chemicals were purchased from Beijing Chemical Reagents Co (Beijing, China). Deionized water was produced by a Milli Q Water Purification system from Millipore (MA, USA), and Fluoro-Jade® B (FJB) was also purchased from Millipore.
Animal models, drug administration and treatment
Adult male Sprague–Dawley (SD) rats (280–300 g, certificate No SCXK2012-0001) were purchased from the Vital River Laboratory Animal Technology Co, Ltd (Beijing, China) and housed under a temperature of 21–24 °C with a 12 h light/dark cycle and a relative humidity of 50%. All animal tests and experimental procedures were reviewed and approved by the Institutional Animal Care and Use Committee of the Chinese Academy of Medical Sciences and Peking Union Medical College.
Rats were anesthetized with 10% chloral hydrate (380 mg/kg), and the right common carotid artery (CCA), internal carotid artery (ICA) and external carotid artery (ECA) were isolated and ligated. To induce cerebral ischemia, a monofilament nylon suture (diameter 0.2 mm) was introduced from CCA into ICA and advanced to middle cerebral artery (MCA), and 1.5 h later, the filament was withdrawn to initiate reperfusion. The body temperature of animals was maintained at 37 °C throughout the procedure.
Animals were randomly divided into six groups: sham (n=40), vehicle (n=40), DGMI (1, 3 and 10 mg/kg, n=40 for each group) and edaravone (5 mg/kg, n=20). Edaravone was used as a free radical scavenger for a positive control. Rats were intravenously treated with vehicle, DGMI or edaravone at the onset of reperfusion and 12 h after the reperfusion.
Measurement of neurological deficit and cerebral infarct volume
Neurological tests were performed by an investigator blinded to the experimental design as described before28. Neurological deficits were scored on a five-point scale: 0: no neurological deficit; 1: failure to extend left paw fully; 2: circling to the left; 3: falling to the left; and 4: unable to walk spontaneously with depressed levels of consciousness. After I/R injury, rats with a score of 0 or 4 were not used for further study. After exclusions, there were 40 rats in the sham group, 36 rats in the vehicle group, 37 rats in the DGMI (1 mg/kg) group, 38 rats in the DGMI (3 and 10 mg/kg) groups, and 19 rats in the edaravone group used for further study.
Neurological deficit was also assessed by tracking and analyzing rat movement behavior. Briefly, rats were placed in a square box (50 cm×50 cm×50 cm) for 10 min. The movement route of each rat over 10 min was tracked and recorded by a camera above the box. The recorded videos were further analyzed by animal behavior analysis software (Shanghai Xinruan Information Technology Co Ltd, SuperMaze V2.0, Shanghai, China).
After assessment of neurological deficit, rats were sacrificed, and brains were quickly removed and dissected into two halves along the sagittal suture. Ischemic and normal hemispheres were dried overnight at 100°C in a drying oven, and weights, both before and after drying, were obtained. Brain water content (%) was calculated as (wet weight - dry weight)/wet weight×100%. Cerebral edema (%) = (ischemic brain water content - normal brain water content)/normal brain water content×100%.
For infarct volume assessment, rats were sacrificed, and the brains were rapidly removed and frozen at -20 °C for 15 min before being dissected into 2 mm coronal sections. Brain sections were then stained with 0.5% TTC for 20 min at 37 °C and fixed with 4% paraformaldehyde. Infarcted tissues remained unstained (white), whereas normal tissues stained red. Brains were photographed, and the infarct areas were calculated by an investigator blinded to the experimental groups using NIH ImageJ software (National Institutes of Health, Bethesda, MD, USA). Infarct volumes were obtained by multiplying brain slice thickness (2 mm) by the infarct area. Results were expressed as a percentage of total hemisphere volume.
Immunofluorescence assay
The ischemic penumbra of the cortex in I/R injury rats or the corresponding cortex region in control rats was used for immunofluorescence assay. For TUNEL and NeuN double labeling, brain tissue slices were incubated in proteinase K solution for 30 min at room temperature. After the brain slices were washed with PBS three times, the buffer solution was added and incubated at room temperature for 20 min. Then, the reaction solution was added and incubated for 60 min. Next, 2×saline sodium citrate was added and incubated for 15 min. After washed with PBS three times, slices were placed in 0.5% Triton X-100 solution for 20 min, after which 10% goat serum was added and incubated for 30 min. Then, primary antibody for NeuN was added and incubated overnight. After the brain slices were washed with PBS three times, the secondary antibody was added and incubated at room temperature for 1 h. Then, DAPI was added and incubated for 10 min. Immunofluorescence images were acquired using a fluorescence microscope.
For FJB and NeuN double labeling, brain tissue was first incubated in 0.0001% FJB dye (containing 0.1% acetic acid) for 30 min at room temperature and then rinsed three times with PBS. Sections were then incubated for 20 min in 0.5% Triton X-100 solution, followed by incubation in 10% goat serum for 30 min. The primary antibody for NeuN was then added and incubated overnight. After the brain slices were washed with PBS three times, the secondary antibody was added and incubated at room temperature for 1 h. Then, DAPI was added and incubated for 10 min. Immunofluorescence images were acquired using a fluorescence microscope.
For Nrf2 or p-CREB double-labeled staining, the slices were placed in 0.5% Triton X-100 solution for 20 min, followed by 10% goat serum for 30 min. Then, primary antibodies for Nrf2, p-CREB and NeuN were added and incubated overnight. After the brain slices were washed with PBS three times, secondary antibodies were added and incubated at room temperature for 1 h. Then, DAPI was added and incubated for 10 min. Immunofluorescence images were acquired using a fluorescence microscope.
PC12 cell culture and OGD/R
PC12 cells were purchased from China Infrastructure of Cell Line Resources, Chinese Academy of Medical Sciences. Cells were cultured in polystyrene flasks or 96-well plates with RPMI-1640 medium containing 5% fetal bovine serum, 10% horse serum, 100 U/mL penicillin and 100 μg/mL streptomycin under a humidified atmosphere of 95% air and 5% CO2. Cell culture medium was carefully replaced every 48 h. To initiate OGD, PC12 cells were first washed with glucose-free RPMI-1640 and then incubated in glucose-free medium in a Thermo Scientific Series 8000 WJ/IR/N2 oxygen-free incubator (94% N2, 5% CO2 and 1% O2) for 4 h. After hypoxia, cells were incubated with full culture medium in the absence or presence of ginkgolide A, B or C or DGMI for 24 h. N-Acetyl-L-cysteine (NAC) was used as a positive control.
Assay for cell viability
Following the above cell treatment protocol, cell viability was assessed by MTT assay. Briefly, cells were seeded in 96-well plates with a density of 8×103 cells per well. MTT reagent was added to each well at a final concentration of 0.5 mg/mL and incubated at 37°C for 4 h. Afterward, medium was carefully removed, and 100 μL DMSO was added and stirred for 15 min on a microtiter plate shaker to dissolve the formazan product. Absorbance was measured at 570 nm using a Molecular Devices SpectraMax M5 microplate reader. Cell viability was expressed as a percentage of the untreated control.
Immunocytochemistry assay
Nrf2 and p-CREB expression was detected by immunofluorescence assay and quantified using the Cellomics ArrayScan VTI high-content analysis platform. PC12 cells were cultured in black-walled optically clear-bottomed 96-well plates (Corning Life Sciences, Acton, MA, USA). Cells were fixed with 4% paraformaldehyde for 15 min following permeabilization with 0.3% Triton X-100 treatment for 10 min. Cells were blocked with 3% BSA for 1 h at 37 °C and stained with primary antibody (1:200) overnight at 4 °C. After washing three times, cells were incubated with secondary antibody conjugated to Alexa Fluor-488 or Alexa Fluor-594 (1:500) for 1 h and Hoechst 33258 for 30 min at 37 °C. Fluorescence images were acquired using the Cellomics ArrayScan VTI HCS Reader.
Western blot analysis
For Western blot analysis, the ischemic penumbra of cortical regions in I/R injury rats and the corresponding cortical regions in controls were used. Total protein was extracted using a Total Protein Extraction Kit following the manufacturers' protocols at 24 h, and nuclear protein was extracted using a Nuclear and Cytoplasmic Protein Extraction Kit. Protein concentration of the supernatants was quantified using a BCA Protein Assay Kit. Proteins were separated by electrophoresis on sodium dodecyl sulfate-polyacrylamide gels (SDS–PAGE). After transferring to PVDF membranes, the membrane was blocked with 5% BSA in TBST for 2 h and incubated overnight at 4 °C with primary antibody for cleaved caspase 3, Bcl-2, Bax, p-Akt, Akt, Nrf2, HO-1, CREB, p-CREB, histone H3 and GAPDH. After the membrane was washed three times in TBST, antibody recognition was performed with the respective secondary antibody against HRP conjugated rabbit or mouse (1:1000 dilution) IgG for 1.5 h at room temperature. Antibody-bound proteins were detected by an enhanced chemiluminescence (ECL) assay and captured through a ChemiDoc-it2 Imager (UVP, Upland, CA, USA). The optical density of the bands was analyzed using NIH ImageJ software.
Statistical analysis
All data are presented as the mean±standard deviation. Statistical analysis was carried out using the SPSS statistical package (Version 16.0; SPSS, Chicago, IL, USA) program, and the significance of each group was verified with one-way analysis of variance (ANOVA) followed by Tukey's multiple comparison post hoc test. A P value <0.05 was considered significant.
Results
DGMI improves neurological deficit, attenuates cerebral edema and reduces infarct volume in I/R injury rats
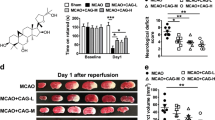
Neurological deficit scores were assessed after 24 h of reperfusion to determine the protective effects of DGMI. As shown in Figure 2A, rats in the I/R group showed severe neurological deficits compared to the sham group (P<0.01). The DGMI 1, 3 and 10 mg/kg groups exhibited dose-dependent decreased neurological deficit scores (P<0.05, P<0.01). The positive control, edaravone, significantly decreased the neurological deficit scores as well (P<0.01). The recorded typical movement route of rats also showed high neurological deficit scores in the I/R group, while low neurological deficit scores were observed in the DGMI treatment groups (Figure 2C).

Neuroprotection effects of DGMI on rat I/R injury. (A) Neurological deficit scores at 24 h after I/R (n=8). (B) Percent of water content (cerebral edema, n=6). (C) The recorded typical movement routes of rats in a box within 10 min. (D) TTC-stained coronal brain slices arranged in order. The white area represented infarcted tissue. (E) Infarct volumes calculated from the slices thickness and infract areas of TTC staining results (n=6). Data were expressed as mean±SD. * P<0.05, ** P<0.01 compared to the vehicle group. ## P<0.01 compared to the sham group.
Water content was examined to study the protective effects of DGMI on cerebral edema. There was no cerebral edema in the sham group (Figure 2B). Compared to the sham group, the percentage of water content in the I/R group was significantly higher (P<0.01). The DGMI 3 and 10 mg/kg treatment groups exhibited significantly reduced water content compared to the I/R group (P<0.05 and P<0.01).
The extent of brain infarctions was assessed by TTC staining. As shown in Figure 2D and Figure 2E, the sham group showed no infarct, while the I/R group showed obvious infarct volume (P<0.01). Infarct volume was significantly decreased after treatment with DGMI at 3 or 10 mg/kg (P<0.05, P<0.01). The positive control, edaravone, also significantly reduced the infarct volume (P<0.01).
DGMI inhibits brain cell apoptosis in I/R injury rats
Cleaved caspase 3 protein was significantly upregulated in the I/R group compared to the sham group (Figure 3A), an effect that was attenuated by treatment with 3 or 10 mg/kg DGMI (P<0.01). As shown in Figure 3B, the ratio of Bax/Bcl-2 in the I/R group was significantly higher than that in the sham group. When treated with 3 or 10 mg/kg DGMI, the ratio was significantly reduced (P<0.05, P<0.01). TUNEL and FJB staining represent the rate of apoptosis. It was found that I/R injury increased the apoptosis rate of neurons, while DGMI significantly downregulated the apoptosis rate of neurons compared to the I/R group (Figure 3C, 3D, 3E and 3F). These results illustrate that DGMI significantly inhibits apoptosis induced by I/R.

Effects of DGMI on cell apoptosis in I/R rats. (A) The expression of cleaved caspase 3. (B) The expression of Bax and Bcl-2. Proteins were prepared and analyzed by Western blot analysis, which were normalized to GAPDH expression (n=6). (C) The TUNEL and NeuN double stained positive cells were shown by immunofluorescence method. (D) The FJB and NeuN double stained positive cells were shown by immunofluorescence method. (E) The percent of TUNEL and NeuN double stained positive cells (n=5). (F) The percent of FJB and NeuN double stained positive cells (n=5). * P<0.05, ** P<0.01 compared to the vehicle group. ## P<0.01 compared to the sham group, Scale bar=50 μm.
DGMI activated the Akt-Nrf2/CREB signaling pathway
Nrf2 is a key transcription factor that regulates expression of antioxidant genes, including HO-1, in response to oxidative stress. CREB is also an important nuclear transcription factor that triggers expression of neuro-protective proteins, including Bcl-2. As shown in Figure 4A, the 3 and 10 mg/kg DGMI groups showed significantly increased p-Akt expression compared to the I/R group (P<0.05, P<0.01). In Figure 4B, Nrf2 protein was significantly upregulated in the DGMI 1, 3 and 10 mg/kg treatment groups compared with the I/R group (P<0.05, P<0.01). As a downstream protein of Nrf2, HO-1 was also significantly upregulated in the DGMI 3 and 10 mg/kg groups compared with the I/R group (Figure 4C, P<0.01). In Figure 4D, the p-CREB protein exhibited relatively low expression in the sham and I/R groups, while the DGMI 3 and 10 mg/kg groups exhibited significantly upregulated p-CREB protein expression compared with the I/R group (P<0.01). Immunofluorescence staining showed that there were little or no Nrf2/NeuN double stained positive cells in the sham and I/R groups, while the numbers of Nrf2/NeuN double stained positive cells were significantly increased in the DGMI treatment groups (Figure 5A and 5B, P<0.01). In addition, few p-CREB/NeuN double stained positive cells were observed in the sham and I/R groups, while the DGMI treatment groups exhibited significantly increased numbers of p-CREB/NeuN double stained positive cells (Figure 5C and 5D, P<0.01).

Effects of DGMI on Akt-Nrf2 and Akt/CREB signaling pathways related protein expression. (A) p-Akt/Akt (n=6). (B) Nrf2 (n=6). (C) HO-1 (n=6). (D) p-CREB/CREB (n=6). Proteins were prepared and analyzed by Western blot analysis, which were normalized to GAPDH or Histone H3 expression. * P<0.05, ** P<0.01 compared to the vehicle group.

Immunofluorescence study of Nrf2 and p-CREB on ischemic penumbra of cortex region in I/R injury rats or the corresponding cortex region in sham rats. (A) The Nrf2 and NeuN double stained positive cells were shown by immunofluorescence method. (B) The number of Nrf2 and NeuN double stained positive cells (n=5). (C) The p-CREB and NeuN double stained positive cells were shown by immunofluorescence method. (D) The number of p-CREB and NeuN double stained positive cells (n=5). ** P<0.01 compared to the model group, Scale bar=50 μm.
Effect of DGMI and ginkgolides A, B and C on OGD/R-induced injury in PC12 cells
As shown in Figure 6, OGD/R caused severe cell injury and induced cell death, while ginkgolide A, B or C treatment protected against cellular injury at a 10 μmol/L concentration (Figure 6A, P<0.01). Meanwhile, DGMI (10 and 20 μg/mL) significantly increased cell viability (Figure 6B, P<0.05 and P<0.01). As a scavenger of ROS, the positive control NAC also showed protective effects on PC12 cells (P<0.01).

Effects of Ginkgolide A, B, C and DGMI on cell viability in PC12 cell after OGD/R. (A) Ginkgolide A, B and C. (B) DGMI (n=10). * P<0.05, ** P<0.01 compared to the model group. ## P<0.01 compared to the normal group.
DGMI and ginkgolides A, B and C protected PC12 cells from OGD/R-induced apoptosis
Next, we investigated the anti-apoptotic effects of DGMI and the major components of DGMI (ginkgolides A, B and C) on OGD/R injury in PC12 cells. As shown in Figure 7A, cleaved caspase 3 protein was significantly upregulated in the OGD/R group compared to the control group, while the DGMI 20 μg/mL treatment reduced cleaved caspase 3 expression significantly (P<0.05). Next, expression of the pro-apoptotic protein Bax and the anti-apoptotic protein Bcl-2 was investigated. As shown in Figure 7B, the ratio of Bax/Bcl-2 in the OGD/R group was significantly increased compared to the control group, while the DGMI 10 and 20 μg/mL treatments significantly reduced the ratio of Bax/Bcl-2 (P<0.05, P<0.01). These results further show that the ginkgolide A, B or C 10 μmol/L groups all significantly reduced cleaved caspase 3 expression (P<0.01, Figure 7C). Ginkgolide A, B or C treatment also significantly reduced the ratio of Bax/Bcl-2 (P<0.01, Figure 7D). These results indicate that ginkgolide A, B or C significantly inhibits apoptosis induced by OGD/R.

Effects of DGMI and Ginkgolide A, B, C on apoptosis-related protein expression in PC12 cell after OGD/R. (A) Effects of DGMI on cleaved caspase 3 expression (n=4). (B) Effects of DGMI on Bax and Bcl-2 expression (n=4). (C) Effects of Ginkgolide A, B and C on cleaved caspase 3 expression (n=6). (D) Effects of Ginkgolide A, B and C on Bax and Bcl-2 expression (n=6). Proteins were prepared and analyzed by Western blot analysis, which were normalized to GAPDH or Histone H3 expression (n=6). * P<0.05, ** P<0.01 compared to the model group. ## P<0.01 compared to the normal group.
Effects of DGMI with or without LY294002 on p-Akt, Nrf2, p-CREB, Bax and Bcl-2 proteins in PC12 cells exposed to OGD/R
As shown in Figure 8A, the Akt phosphorylation level was relatively low in the normal and OGD/R groups, while DGMI treatment significantly upregulated phosphorylation of Akt (P<0.01), and the PI3K/Akt inhibitor LY294002 reversed this phenomenon (P<0.01). Next, nuclear Nrf2 levels were upregulated in the DGMI group (P<0.01), and this effect could be reversed by treatment with LY294002 (P<0.01, Figure 8B). As shown in Figure 8C, the p-CREB level was relatively low in the normal and OGD/R groups, while DGMI treatment significantly up-regulated the phosphorylation level of CREB (P<0.01), and LY294002 reversed this induction (P<0.01). Moreover, LY294002 also reversed the downregulation of the Bax/Bcl-2 ratio induced by DGMI (P<0.01). The above results illustrate that the protective effect of DGMI may be related to regulation of the PI3K/Akt pathway.

Effects of DGMI with or without LY294002 on p-Akt, Nrf2, p-CREB, Bax and Bcl-2 proteins in PC12 cells exposed to OGD/R. (A) pAkt/Akt. (B) Nrf2. (C) p-CREB/CREB. (D) Bax/Bcl-2. Proteins were prepared and analyzed by Western blot analysis, which were normalized to GAPDH or Histone H3 expression (n=4). * P<0.05, ** P<0.01 compared to the model group. ## P<0.01 compared to the normal group. && P<0.01 compared to the DGMI group.
Ginkgolides A, B and C activated Akt-Nrf2 and Akt/CREB pathways in PC12 cells exposed to OGD/R
In Figure 9, Western blot analysis shows that treatment with 10 μmol/L ginkgolide A, B or C significantly increased phosphorylation of Akt, expression of Nrf2, HO-1, and phosphorylation of CREB, which was suppressed by LY294002. Immunofluorescence results also revealed that 10 μmol/L ginkgolide A, B or C significantly increased nuclear Nrf2 and p-CREB levels, which was reversed by the addition of LY294002 (Figure 10). These results suggest that ginkgolide A, B or C can all activate Nrf2 and p-CREB in PC12 cells exposed to OGD/R and that this effect is PI3K/Akt dependent.

Effects of Ginkgolide A, B and C on p-Akt, Nrf2, HO-1 and p-CREB proteins expression. (A) pAkt/Akt (n=6). (B) Nrf2 (n=6). (C) HO-1 (n=6). (D) p-CREB/CREB (n=4). Proteins were prepared and analyzed by Western blot analysis, which were normalized to GAPDH or Histone H3 expression. ** P<0.01 compared to the model group. && P<0.01 compared to the related group linked by lines.

Effects of Ginkgolide A, B and C on Nrf2 and p-CREB levels. (A) Expression of Nrf2 (green fluorescence) was assessed by immunofluorescence in PC12 cells. (B) Expression of p-CREB (red fluorescence) was assessed by immunofluorescence in PC12 cells. (C) Mean fluorescence intensity of Nrf2 (n=5). (D) Mean fluorescence intensity of p-CREB (n=5). ** P<0.01 compared to the model group. && P<0.01 compared to the related group linked by lines.
Discussion
In the present study, DGMI treatment significantly reduced infarct volumes and neurological deficit scores at all dosages employed. Moreover, DGMI significantly inhibited apoptosis in neurons, activated PI3K/Akt signaling, and mediated the activation of Nrf2 and CREB. The PI3K/Akt pathway is one of the most important pathways involved in the regulation of cell survival, proliferation and differentiation that can be activated by various stimuli, including growth factors, hormones, medical drugs and xenobiotics. Nrf2 and CREB are downstream targets of the PI3K/Akt pathway, and we observed that activation of Akt signaling could trigger nuclear translocation of Nrf2 and p-CREB to promote cell survival and inhibit apoptosis4,12.
Ischemia reperfusion injury immediately and acutely induces the production of intracellular ROS, causing an imbalance between oxidants and antioxidants in the brain. The damaged antioxidant defense system causes further cell dysfunction, apoptosis, autophagy and even death. Fortunately, this defense system can be maintained via the intake and self-production of antioxidants by the cell. Increasing the level of antioxidants is a promising strategy for the treatment of ischemic stroke. Among various regulation factors, Nrf2 and HO-1 have been suggested to be effective in promoting antioxidant production29. Recent studies demonstrated that HO-1 expression is significantly increased in both neuronal and non-neuronal cells after cerebral ischemia reperfusion30,31,32. Up-regulation of HO-1 has been shown to be protective against neuronal injury induced by oxygen-glucose deprivation33. In addition, Nrf2 activator has been shown to ameliorate hemorrhagic transformation in focal cerebral ischemia by protecting endothelial cells and pericytes and upregulating expression of tight junction proteins34. In our study, the Nrf2 protein was significantly upregulated in the DGMI treatment groups compared with the I/R group in vivo. In addition, HO-1, as one of its downstream proteins, was also significantly upregulated in the DGMI treatment groups. The results of immunofluorescence staining showed that the numbers of Nrf2/NeuN double stained positive cells were significantly increased in the DGMI treatment groups.
CREB was reported to be an effective activator of Bcl-27. As an anti-apoptotic protein, Bcl-2 inhibits intracellular ROS production and suppresses pro-apoptotic protein expression. CREB is activated by the phosphorylation of other kinases, one of which is Akt4. Once activated, CREB protein, together with other transcriptional coactivators, binds the CRE promoter to facilitate transcription of various genes, including Bcl-27. Upregulation of Bcl-2 by CREB activation suppressed activation of caspases, protecting the cells from apoptosis. In our study, the DGMI treatment groups exhibited significantly upregulated expression of p-CREB protein compared with the I/R group. The DGMI treatment groups also exhibited significantly increased numbers of p-CREB/NeuN double stained positive cells in the ischemic penumbra of cortical regions.
Ginkgolides A, B and C are the major components of DGMI, and our study demonstrated that ginkgolides A, B and C reversed OGD/R-induced PC12 cell injury. In addition, ginkgolide A, B and C treatment led to an increase in the levels of Akt phosphorylation and expression of Nrf2. The accumulation of Nrf2 in the nucleus induced expression of the downstream antioxidant enzyme HO-1. CREB was also activated by ginkgolide A, B and C treatment. Interestingly, activation of Nrf2 and CREB and the neuroprotective effects of ginkgolides A, B and C were markedly attenuated when the expression of the PI3K/Nrf2 pathway was inhibited using LY294002, a PI3K inhibitor. Taken together, these in vitro findings supported our hypothesis that ginkgolides A, B and C protected against OGD/R-induced PC12 cell injury via activation of Nrf2 and CREB through PI3K/Akt signaling (Figure 11).

Proposed mechanisms for the protective effects of DGMI against ischemia/reperfusion damage. DGMI and its components Ginkgolide A, B and C upregulated PI3K/Akt signaling, which induced Nrf2 and CREB activation. Further, Nrf2 nuclear translocation enhances the expression of HO-1, and CREB activation upregulated Bcl-2 expression. Finally, HO-1 and Bcl-2 protects cells against apoptosis.
In summary, this study demonstrates that DGMI ameliorates I/R injury in rats by activating the PI3K/Akt-mediated Nrf2 and CREB signaling pathway. Its major components, ginkgolides A, B and C, contributed to these protective effects, and these ginkgolides might also be developed into therapies for the treatment of cerebral ischemia-induced brain damage.
Author contribution
The paper was designed, performed and written by Wen ZHANG. Jun-ke SONG and Rong YAN performed the animal experiments and analyzed the data. Li LI performed the cell experiments. Zhi-yong XIAO, Wen-xia ZHOU, Zhen-zhong WANG and Wei XIAO contributed reagents and gave advices on signaling pathway studies and animal model studies. Guan-hua DU, the corresponding author of this paper, designed the work and led the research. All the listed authors have read and approved the submitted manuscript.
Abbreviations
Akt, protein kinase B; ARE, antioxidant response element; bZip, basic leucine zipper; CBP, CREB-binding protein; CCA, common carotid artery; CNC, Cap'n'Collar; CREB, cyclic AMP-responsive element binding protein; DGMI, diterpene ginkgolides meglumine injection; DMSO, dimethyl sulfoxide; ECA, external carotid artery; FJB, Fluoro-Jade® B; HO-1, heme oxygenase-1; ICA, internal carotid artery; I/R, ischemia/reperfusion; Keap1, kelch-like ECH-associated protein-1; Maf, musculoaponeurotic fibrosarcoma; MCA, middle cerebral artery; MCAO/R, middle cerebral artery occlusion and reperfusion; MMP, mitochondrial membrane potential; MTT, 3-[4,5-dimethyl-2-thiazolyl]-2,5-diphenyl-2H-tetrazolium bromide; NOX2, NADPH oxidase 2; Nrf2, nuclear factor-erythroid 2-related factor 2; OGD/R, oxygen-glucose deprivation/reperfusion; PI3K, phosphoinositide-3-kinase; ROS, reactive oxygen species; SD, Sprague–Dawley.
References
Moretti A, Ferrari F, Villa RF. Neuroprotection for ischaemic stroke: current status and challenges. Pharmacol Ther 2015; 146: 23–34.
Hu M, Liu ZJ, Lv PY, Wang HB, Zhu YF, Qi QQ, et al. Autophagy and Akt/CREB signalling play an important role in the neuroprotective effect of nimodipine in a rat model of vascular dementia. Behav Brain Res 2017; 325: 79–86.
Jia N, Sun QR, Su Q, Dang SK, Chen GM. Taurine promotes cognitive function in prenatally stressed juvenile rats via activating the Akt-CREB-PGC1alpha pathway. Redox Biol 2016; 10: 179–90.
Wang YL, Zhang JJ, Han M, Liu B, Gao YL, Ma P, et al. SMND-309 promotes neuron survival through the activation of the PI3K/Akt/CREB-signalling pathway. Pharm Biol 2016; 54: 1982–90.
Yan TT, Zhao Y, Zhang X, Lin XT. Astaxanthin inhibits acetaldehyde-induced cytotoxicity in SH-SY5Y cells by modulating Akt/CREB and p38MAPK/ERK signaling pathways. Mar Drugs 2016; 14: 56.
Cheng CY, Tang NY, Kao ST, Hsieh CL. Ferulic acid administered at various time points protects against cerebral infarction by activating p38 MAPK/p90RSK/CREB/Bcl-2 anti-apoptotic signaling in the subacute phase of cerebral ischemia-reperfusion injury in rats. PLoS One 2016; 11: e0155748.
Shi RF, Yuan KX, Hu B, Sang HF, Zhou LZ, Xie Y, et al. Tissue kallikrein alleviates cerebral ischemia-reperfusion injury by activating the B2R-ERK1/2-CREB-Bcl-2 signaling pathway in diabetic rats. Oxid Med Cell Longev 2016; 2016: 1843201.
Unoki T, Abiko Y, Toyama T, Uehara T, Tsuboi K, Nishida M, et al. Methylmercury, an environmental electrophile capable of activation and disruption of the Akt/CREB/Bcl-2 signal transduction pathway in SH-SY5Y cells. Sci Rep 2016; 6: 28944.
Buendia I, Michalska P, Navarro E, Gameiro I, Egea J, León R. Nrf2-ARE pathway: an emerging target against oxidative stress and neuroinflammation in neurodegenerative diseases. Pharmacol Ther 2016; 157: 84–104.
Alam J, Stewart D, Touchard C, Boinapally S, Choi AMK, Cook JL. Nrf2, a Cap'n'Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J Biol Chem 1999; 274: 26071–8.
Chan K, Lu R, Chang JC, Kan YW. NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc Natl Acad Sci U S A 1996; 93: 13943–8.
Li Z, Ma QQ, Yan Y, Xu FD, Zhang XY, Zhou WQ, et al. Edaravone attenuates hippocampal damage in an infant mouse model of pneumococcal meningitis by reducing HMGB1 and iNOS expression via the Nrf2/HO-1 pathway. Acta Pharmacol Sin 2016; 37: 1298–306.
Amersi F, Buelow R, Kato H, Ke B, Coito AJ, Shen XD, et al. Upregulation of heme oxygenase-1 protects genetically fat Zucker rat livers from ischemia/reperfusion injury. J Clin Invest 1999; 104: 1631–9.
Saleem S, Zhuang H, Biswal S, Christen Y, Doré S. Ginkgo biloba extract neuroprotective action is dependent on heme oxygenase 1 in ischemic reperfusion brain injury. Stroke 2008; 39: 3389–96.
Alfieri A, Srivastava S, Siow RCM, Cash D, Modo M, Duchen MR, et al. Sulforaphane preconditioning of the Nrf2/HO-1 defense pathway protects the cerebral vasculature against blood–brain barrier disruption and neurological deficits in stroke. Free Radic Biol Med 2013; 65: 1012–22.
Peng B, Zhao P, Lu YP, Chen MM, Sun H, Wu XM, et al. Z-ligustilide activates the Nrf2/HO-1 pathway and protects against cerebral ischemia–reperfusion injury in vivo and in vitro. Brain Res 2013; 1520: 168–77.
Panahian N, Yoshiura M, Maines MD. Overexpression of heme oxygenase-1 is neuroprotective in a model of permanent middle cerebral artery occlusion in transgenic mice. J Neurochem 1999; 72: 1187–203.
Li SZ. Compendium of Materia Medica. People's Medical Publishing House 1982; Beijing.
Xie ZW. Encyclopedia of Chinese herbal medicines. People's Medical Publishing House 1997; Beijing.
Oyama Y, Chikahisa L, Ueha T, Kanemaru K, Noda K. Ginkgo biloba extract protects brain neurons against oxidative stress induced by hydrogen peroxide. Brain Res 1996; 712: 349–52.
Tulsulkar J, Shah ZA. Ginkgo biloba prevents transient global ischemia-induced delayed hippocampal neuronal death through antioxidant and anti-inflammatory mechanism. Neurochem Int 2013; 62: 189–97.
Zhang ZJ, Peng DT, Zhu HY, Wang XM. Experimental evidence of Ginkgo biloba extract EGB as a neuroprotective agent in ischemia stroke rats. Brain Res Bull 2012; 87: 193–8.
Nakanishi K. The ginkgolides. Pure Appl Chem 1997; 14: 89–113.
Qin XF, Lu XJ, Ge JB, Xu HZ, Qin HD, Xu F. Ginkgolide B prevents cathepsin-mediated cell death following cerebral ischemia/reperfusion injury. Neuroreport 2014; 25: 267–73.
Pei HX, Hua R, Guan CX, Fang X. Ginkgolide B reduces the degradation of membrane phospholipids to prevent ischemia/reperfusion myocardial injury in rats. Pharmacology 2015; 96: 233–9.
Ma WJ, Hu J, Cheng Y, Wang JL, Zhang XT, Xu M. Ginkgolide B protects against cisplatin-induced ototoxicity: enhancement of Akt-Nrf2-HO-1 signaling and reduction of NADPH oxidase. Cancer Chemother Pharmacol 2015; 75: 949–59.
Wu QL, Sun YC, Wan Q, Wang ZZ, Bi YA, Wu XY, et al. Determination of ginkgolide A, B, C, and K in diterpene lactones from Ginkgo biloba for injection by HPLC-ELSD. Drugs Clin 2014; 29: 255–8.
Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 1989; 20: 84–91.
Huang JY, Yuan YH, Yan JQ, Wang YN, Chu SF, Zhu CG, et al. 20C, a bibenzyl compound isolated fromGastrodia elata, protects PC12 cells against rotenone-induced apoptosis via activation of the Nrf2/ARE/HO-1 signaling pathway. Acta Pharmacol Sin 2016; 37: 731–40.
Empty
Gao YX, Xu XJ, Chang S, Wang YJ, Xu YZ, Ran SQ, et al. Totarol prevents neuronal injury in vitro and ameliorates brain ischemic stroke: Potential roles of Akt activation and HO-1 induction. Toxicol Appl Pharmacol 2015; 289: 142–54.
Yang PS, Lin PY, Chang CC, Yu MC, Yen TL, Lan CC, Jayakumar T, et al. Antrodia camphorata potentiates neuroprotection against cerebral ischemia in rats via downregulation of iNOS/HO-1/Bax and activated caspase-3 and inhibition of hydroxyl radical formation. Evid Based Complement Alternat Med 2015; 2015: 232789.
Li QF, Zhu YS, Jiang H, Xu H, Liu HP. Up-regulation of heme oxygenase-1 by isoflurane preconditioning during tolerance against neuronal injury induced by oxygen glucose deprivation. Acta Biochim Biophys Sin 2008; 40: 803–10.
Imai T, Takagi T, Kitashoji A, Yamauchi K, Shimazawa M, Hara H. Nrf2 activator ameliorates hemorrhagic transformation in focal cerebral ischemia under warfarin anticoagulation. Neurobiol Dis 2016; 89: 136–46.
Acknowledgements
This work was supported by grants from the National Science and Technology Major Projects for “Major New Drugs Innovation and Development” (No 2013ZX09402203); the National Natural Science Foundation of China (No 81603100); the Graduate Student Innovation Fund of the Chinese Academy of Medical Sciences and Peking Union Medical College (No 2015-1007-07); the Open Project of the State Key Laboratory of Toxicology and Medical Countermeasures (No TMC201510); and The CAMS Innovation Fund for Medical Sciences (No 2017-I2M-1-010).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Zhang, W., Song, Jk., Yan, R. et al. Diterpene ginkgolides protect against cerebral ischemia/reperfusion damage in rats by activating Nrf2 and CREB through PI3K/Akt signaling. Acta Pharmacol Sin 39, 1259–1272 (2018). https://doi.org/10.1038/aps.2017.149
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2017.149
Keywords
This article is cited by
-
NRF2 activation ameliorates blood–brain barrier injury after cerebral ischemic stroke by regulating ferroptosis and inflammation
Scientific Reports (2024)
-
Efficacy of ginkgo diterpene lactone meglumine on cognitive function in patients with acute ischemic stroke: a predefined exploratory analysis of a multicenter, double-blind, randomized controlled trial
Journal of Neurology (2024)
-
Study on correlations of BDNF, PI3K, AKT and CREB levels with depressive emotion and impulsive behaviors in drug-naïve patients with first-episode schizophrenia
BMC Psychiatry (2023)
-
Schisandrin B Protects against Ischemic Brain Damage by Regulating PI3K/AKT Signaling in Rats
Chinese Journal of Integrative Medicine (2023)
-
A novel CREB5/TOP1MT axis confers cisplatin resistance through inhibiting mitochondrial apoptosis in head and neck squamous cell carcinoma
BMC Medicine (2022)
