
Abstract
Alternatively activated macrophages (AAMs) are not only associated with asthma but also lead to asthmatic airway inflammation and remodeling. Inhibition of AAMs is an alternative therapeutic strategy for treating asthma. In this study we investigated whether emodin (1,3,8-trihydroxy-6-methylanthraquinone), isolated from the rhizome of Rheum palmatum, alleviated asthmatic airway inflammation and reduced AAM polarization in a murine asthma model. Mice were sensitized with a triple allergen mix containing dust mite, ragweed and aspergillus (DRA). In mice with DRA-induced asthma, asthmatic inflammation was significantly enhanced. Intraperitoneal injection of emodin (20 mg·kg−1·d−1, ip) 1 h prior to DRA challenge on days 12-14 significantly decreased pulmonary eosinophil and lymphocyte infiltration, mucus secretion and serum IgE production, as well as IL-4 and IL-5 production in bronchoalveolar lavage fluid. In response to emodin treatment, activated markers of AAM Ym-1, Fizz-1 and arginase-1 in the lung tissues were remarkably decreased. In mouse bone marrow-derived macrophages (BMDMs) in vitro, emodin (2–50 μmol/L) dose-dependently inhibited IL-4-induced AAM polarization and STAT6 phosphorylation. Collectively, our results suggest that emodin effectively ameliorates asthmatic airway inflammation and AAM polarization, and it may therefore become a potential agent for the treatment of asthma.
Similar content being viewed by others

Introduction
Asthma is a complex pulmonary disease that affects approximately 300 million people worldwide, from infants to seniors, and leads to 0.25–0.345 million deaths per year1,2. It is a long-term recurring inflammatory disease characterized by inflammatory cell infiltration, reversible airflow obstruction, mucus secretion and bronchospasm3. Because the underlying molecular, cellular and pathogenic mechanisms of this disease have not been fully defined, clinical treatment strategies have been limited to targeting the symptoms of asthma4. Therefore, it is of great importance to develop novel medicines for treating asthma.
As an inflammatory disease, asthma is considered a Th2-dominant disease marked by increased IgE generation, enhanced IL-4 and IL-5 production, mucus secretion, and substantial eosinophil accumulation in the lung lobes5. In response to general antigens such as house dust mites or aspergillus, the adaptive immune system recognizes the antigens presented by the antigen-presenting cells (APCs), activates the Th2 cells, and increases antigen-specific IgE production, which is predominantly modulated by Th2 lymphocytes, dendritic cells, mast cells and eosinophils6,7,8. Recently, macrophages have been identified as additional immune cells that not only are associated with Th2 responses but also contribute to asthmatic inflammation9. Depletion of pulmonary macrophages or the adoptive transfer of alternatively activated macrophages (AAMs) is able to attenuate or accentuate asthmatic airway inflammation10,11. Therefore, restricting Th2-related macrophage polarization may provide an alternative strategy for the clinical treatment of asthma.
Two classes of medicines are generally used to treat asthma. Short-acting, selective beta-2 adrenergic agonists (SABA) are the first-line treatment for acute asthmatic symptoms, whereas corticosteroids and long-acting, selective beta-2 adrenergic agonists (LABA) are the most effective medicines for long-term control12. Although these medicines can ameliorate the symptoms of asthma, complications of treatment may include immunosuppression, microorganism infection, and progressive loss of pulmonary function13. In addition, some asthmatic patients, including those patients with asthma with neutrophilic inflammation, resist the use of the standard corticosteroid therapy14. Therefore, new therapeutic medicines for treating asthma must be developed.
Natural compounds have been widely used for treating inflammatory diseases including allergic diseases. Emodin (1,3,8-trihydroxy-6-methylanthraquinone) can be isolated from the rhizome of Rheum palmatum L, and it has been used to treat inflammation, tumors, and cardiovascular diseases15,16,17. In the present study, we demonstrated that emodin alleviated triple allergen (dust mice, ragweed, and aspergillus; DRA)-induced asthmatic inflammation, including decreased IgE generation, reduced IL-4 and IL-5 production, and attenuated pulmonary eosinophil infiltration. Notably, emodin down-regulated IL-4-mediated macrophage polarization and reduced STAT6 phosphorylation. Thus, these findings suggested that emodin might become a potential candidate agent for treating asthmatic airway inflammation.
Materials and methods
Animals
Male C57BL/6 mice (6 weeks old, 20±2 g, specific pathogen free) were purchased from SLAC Laboratory Animal Corporation (Shanghai, China). Five mice were housed in one cage in a climate-controlled room (25 °C, 55% humidity and 12 h light/dark cycle). The mice were allowed ad libitum access to water and food and were allowed to acclimate for at least 2 weeks prior to the beginning of the specific pathogen-free condition of the experiment18. This research adhered to the Principles of Laboratory Animal Care (NIH publication No 85-23, revised 1996). All experimental protocols described in this study were approved by the Animal Care and Use Committee of Shanghai Jiao Tong University.
Reagents
Emodin was purchased from Sigma (St Louis, MO, USA), and a 50 mmol/L stock solution was prepared in DMSO and stored in small aliquots at -20 °C. The ELISA kits for detecting IL-4 and IL-5 were ordered from R&D systems (Minneapolis, Minnesota, USA). The primers used in this study were synthesized by HuaGene Biotech Co, Ltd (Shanghai, China). The primary antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA), including antibodies against pY-STAT6, total-STAT6, KLF4 and GAPDH. Other chemical reagents without special indications were obtained from Sigma (St Louis, MO, USA).
Allergens
The triple allergens include extracts of dust mite (Dermatophagoides farina), ragweed (Ambrosia artemisiifolia), and Aspergillus fumigates (Greer Laboratories, Lenoir, NC). Aluminum (Inject Alum; Thermo Scientific) was used as an adjuvant. The quantities of allergens for intraperitoneal injection (200 μL) per mouse included: dust mite (5 μg), ragweed (50 μg), and Aspergillus fumigates (5 μg). The quantities of allergens for intranasal administration (50 μL) were as follows: dust mite (8.3 μg), ragweed (83.4 μg), and Aspergillus fumigates (8.3 μg)11.
Murine asthma models
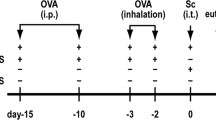
To determine the effect of emodin on murine asthma, we dissolved emodin in vehicle (10% DMSO, 60% cremophor, 20% ethanol and 10% PBS) (final concentration) for the in vivo experiments19. Mice were randomly assigned to one of three treatment groups: the control group (PBS+vehicle), the DRA group (DRA+vehicle) or the emodin treatment group (DRA+emodin). Each group contained five mice. The mice were sensitized by an intraperitoneal (ip) injection of 200 μL alum-precipitated antigens containing the triple allergen mix (60 μg of DRA) in PBS vehicle. Five days after the first immunization (D0), the mice were given a second intraperitoneal booster of the same dose of allergens in alum adjuvant (D5) followed by exposure to DRA (100 μg) combination (50 μL) or PBS (50 μL) as a control on d 12–14 by intranasal (in) administration after anesthesia with pentobarbital sodium. Emodin was administered 1 h prior to DRA administration by intraperitoneal injection on d 12–14. On day 15, the mice were anesthetized, and peripheral blood, bronchoalveolar lavage (BAL) fluid and lung tissues were collected for further analysis.
Collection of bronchoalveolar lavage fluid (BALF) and total cell count
The lungs were lavaged three times with 0.6 mL of PBS via a tracheal catheter, and BAL fluid was centrifuged at 4 °C20. The cell pellets were resuspended in PBS for total cell counts using a hemocytometer. Cell smears were prepared using cytospin (Thermo Electron Corporation, UK) and were stained with Hema-3. Neutrophils, macrophages and lymphocytes were identified via microscopy.
Flow cytometry assay
Cells collected from BAL fluid were incubated with Fc blocking antibody and anti-mouse CD16/32 antibody (BD Bioscience), followed by PE-conjugated anti-Siglec F (BD Bioscience) and APC-conjugated anti-CD11c (Biolegend) antibodies. Cells were detected via flow cytometry (LSRFortessa TM X-20, BD Biosciences, San Jose, CA, USA), and data were analyzed using FlowJo software.
Histopathological analysis
Lung tissues (left lobe) were fixed with 4% paraformaldehyde, embedded in paraffin and cut into 5-μm-thick sections in a microtome (RM2235, Leica Biosystems, Wetzlar, Germany). Sections were stained with hematoxylin and eosin (H&E) and Periodic acid-Schiff (PAS) for analysis of inflammatory goblet cells, and then images were captured by a microscope (RX51, Olympus Optical Co Ltd, Tokyo, Japan). To quantify mucus production, the “Polygen selections” function in ImageJ software was used to draw the outline of the bronchioles, and then we calculated the perimeter of each bronchiole length using pixels as a measurement unit under the same magnification rate. The number of PAS-positive cells in each bronchiole was divided by the bronchiole length. The mucus score was determined by the number of PAS-positive cells per 100 pixel lengths of the bronchiole.
Collection of serum and analysis of IgE
The mice were anesthetized on day 15 (D15), after which the blood was collected and centrifuged at 4 °C (3000 r/min) for 15 min. Next, the serum was used to detect the total IgE level with an ELISA kit (eBioscience) according to the manufacturer's instructions.
Measurement of cytokine production
BALF was collected to measure the expression of cytokines IL-4 and IL-5 using ELISA kits (R&D Systems, Minneapolis, MN, USA). The cytokine concentrations were interpolated from the standard curves for recombinant IL-4 and IL-58.
Preparation of BMDMs
BMDMs were isolated from male C57BL/6 mice and cultured in DMEM supplemented with antibiotics (100 U/mL of penicillin and 100 μg/mL streptomycin), 10% FBS and 10% culture supernatant of L929 cells for 7 d (37 °C, 5% CO2) 21. Cells were treated with the indicated concentrations of emodin according to experiment requirements, and 0.1% DMSO was added into the culture medium as the solvent control.
RNA isolation, reverse transcription and quantitative PCR
Frozen lung tissue (from one of the right upper lobes) was homogenized, and total RNA was isolated using TRIzol Reagent (Invitrogen, Carlsbad, CA, USA)20. For in vitro analysis, BMDMs (5×105) were incubated with emodin (0, 2, 10 and 50 μmol/L) and challenged with IL-4 for 24 h. The cells were collected, and total RNA was isolated as well. cDNA was prepared using a ReverTra Ace qPCR RT Kit (Toyobo, Osaka, Japan) and amplified by real-time PCR on a StepOne Plus (Thermo Fisher Scientific, Waltham, MA, USA) with primer sets for arginase-1 (forward, 5′--3′; reverse, 5′--3′), Ym-1 (forward, 5′--3′; reverse, 5′--3′), Fizz-1 (forward, 5′--3′; reverse, 5′--3′) and GAPDH (forward, 5′--3′; reverse, 5′--3′).
Western blotting analysis
BMDMs were plated in 6-well plates (1.0×106 cells per well) and incubated overnight (37 °C, 5% CO2). Next, the cells were treated with different doses of emodin (0, 2, 10 and 50 μmol/L) for 0.5 h before they were stimulated with IL-4 (10 ng/mL) for either 30 min or 24 h. Cells were collected with loading buffer (175 mmol/L Tris-HCl, 100 mmol/L DTT, 4.0% SDS, 7.5% glycerinum and 0.2% bromophenol blue in ddH2O), and the proteins were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and were subsequently transferred to a polyvinylidene difluoride (PVDF) membrane. The membranes were washed and blocked in 5% bovine serum albumin (BSA) at room temperature for 1 h. The samples were then probed at 4 °C overnight with primary antibodies (1:1000 dilution) as mentioned above, followed by incubation with secondary antibodies (KPL, Gaithersburg, MD, USA) for 1 h at room temperature22. Quantification of Western blots was performed using ImageJ software (National Institute of Mental Health, Bethesda, MD, USA).
Statistical analysis
Data are presented as the mean±SEM and were obtained from at least three independent tests. One-way ANOVA and Student's t-test (paired comparison) were performed using Prism 5 (GraphPad, San Diego, CA, USA). P<0.05 was considered statistically significant.
Results
Emodin dampened the development of DRA-induced asthmatic inflammation
Pulmonary eosinophil infiltration is one characteristic of asthmatic inflammation. To determine whether emodin alleviates asthma, we established a DRA-induced murine asthma model, and the role of emodin in asthma was evaluated (Figure 1A). On day 15, 24 h after the last challenge with DRA, the lungs were lavaged, and the bronchoalveolar lavage fluid (BALF) was collected for the assessment of inflammatory cell accumulation. Based on the flow cytometry assay, compared with the control mice that received vehicle and had no eosinophil accumulation except for alveolar macrophages (Siglec-F+CD11c+), DRA challenge led to 96% eosinophil (Siglec-F+CD11c−) infiltration in BAL cells in the lung. Emodin significantly reduced eosinophil accumulation to 69% in BAL cells (Figure 1B). Then, we performed quantitative analysis and found that DRA remarkably induced total inflammatory cell and eosinophil infiltration, which were dramatically decreased in response to emodin treatment (Figure 1C and D). In addition, emodin also significantly reduced DRA-induced lymphocyte infiltration (Figure 1E). These results indicated that emodin ameliorated pulmonary inflammatory cell and eosinophil infiltration in DRA-induced asthmatic inflammation.

Emodin dampened the development of DRA-induced asthmatic inflammation. (A) The schematic timeline showed that mice were sensitized with DRA on Days 0 and 5 and challenged with DRA on Days 12, 13 and 14. On Day 15, the mice were sacrificed for analysis of the asthmatic inflammation. In the emodin treatment group, the mice received intraperitoneal injections of emodin (20 mg/kg) 1 h prior to DRA administration on days 12, 13 and 14. (B) Infiltrated inflammatory cells in BAL fluid were stained with different markers for identifying eosinophils (Eos, Siglec F+CD11c−) and macrophages (Mac, Siglec F+CD11c+), and then they were analyzed using flow cytometry. Total infiltrated cells (C), eosinophils (D) and lymphocytes (E) in BAL fluid were counted based on the total number of BAL cells and the percentage of different cell types. Values represent mean±SEM. n=5 mice per group, and ND stands for 'not detected'. ** P<0.01 between the indicated groups.
Emodin alleviated eosinophilic lung injury in DRA-induced asthma
After the last DRA or PBS challenge, lung tissues were collected, and histological examinations were performed to determine the extent of eosinophilic lung injury. As shown in Figure 2A, compared with the PBS challenge, DRA induced increased inflammatory cell infiltration into the peribronchiolar and perivascular connective tissues. Moreover, emodin significantly attenuated the eosinophil infiltration. Mucus secretion is another inflammatory feature of allergic asthma, and PAS staining was performed to evaluate airway hypersecretion of mucus and goblet cell hyperplasia. In DRA-challenged mice, increased mucus hypersecretion and goblet cell hyperplasia were detected in the bronchi compared with no PAS-positive cells in the PBS-treated control mice. DRA-induced mucus hypersecretion was significantly diminished in response to emodin treatment (Figure 2B). To further assess the degree of mucous gland and goblet cell hyperplasia, the PAS-positive cells were quantified (Figure 2C). These pathological results demonstrated that emodin inhibited DRA-induced allergic airway inflammation.

Emodin ameliorated asthmatic lung injury in DRA-induced asthma. Lung lobes were excised, fixed and processed for histological evaluation. (A) H&E staining was performed to determine the asthmatic inflammation in DRA-induced asthmatic inflammation in response to vehicle or emodin treatment. The black arrows indicate pulmonary infiltrated eosinophils. (B) PAS staining was performed for the identification of goblet cells in the epithelium. Black arrows indicate PAS-positive goblet cells. (C) The number of PAS-positive cells per 100 pixel lengths of the bronchiole was determined as described in the materials and methods section. The results represent mean±SEM (n=5 mice for each group), and ND stands for 'not detected'. * P<0.05 between the indicated groups.
Emodin inhibited DRA-induced IgE generation and type 2 cytokine production
Asthma has been defined as Th2-mediated airway allergic inflammation, in which the production of IgE, IL-4 and IL-5 is increased in response to allergen challenge. Twenty-four hours after the last stimulation with DRA, IgE in serum and IL-4 and IL-5 in BALF were enhanced (Figure 3). Emodin significantly reduced IgE (Figure 3A), IL-4 (Figure 3B) and IL-5 (Figure 3C) production with inhibition rates of approximately 24.4%, 34.2% and 39.7%, respectively. These data revealed that emodin attenuated asthmatic inflammation and reduced type 2 cytokine production.

Emodin inhibited IgE generation in serum and IL-4 and IL-5 production in BALF. Mouse serum and BALF were collected 24 h after the last DRA challenge. (A) Total IgE production in mouse serum was detected by ELISA. The production of IL-4 (B) and IL-5 (C) was detected by ELISA. Values were presented as the mean±SEM (n=5 mice for each group). ND stands for 'not detected'. * P<0.05 between the indicated groups.
Emodin decreased polarization of AAMs during acute allergic airway inflammation
Type 2 cytokine IL-4 can induce AAMs, and our previous reports have proven that AAMs are not only associated with asthmatic inflammation but also required for asthma development11. Therefore, we determined whether emodin attenuates the polarization of AAMs in vivo in a murine asthma model. Here, we isolated and collected lung tissues and detected the expression of AAM marker genes arginase-1, Ym-1 and Fizz-1. With treatment with DRA, murine lung tissues displayed a 9-fold increase in the amount of arginase-1, a 55-fold increase in Ym-1 and a 65-fold increase in Fizz-1 production compared with PBS-treated control mice. In response to emodin, the induction of these genes was dramatically reduced to less than 4-fold, 12-fold and 20-fold, respectively (Figure 4). These data indicated that acute challenge with DRA induced AAM polarization, and emodin could attenuate the polarization of AAMs in vivo.

Mice receiving emodin treatment displayed impaired AAM polarization during acute allergic airway inflammation. The lung tissues were excised and collected 24 h after the last DRA challenge, the total RNA was isolated from lung homogenates, and the supernatant was stored. The mRNA levels of arginase-1 (A), Ym-1 (B) and Fizz-1 (C) were then quantified by real-time PCR. Values were represented as the mean±SEM. n=5. * P<0.05, ** P<0.01 between the indicated groups.
Emodin inhibited AAM polarization in vitro
Since the expression of AAM markers was attenuated in the emodin-treated asthma model, we investigated whether emodin inhibits AAM polarization in vitro. After bone marrow-derived macrophages (BMDMs) isolated from C57BL/6 mice were stimulated with 10 ng/mL IL-4 for 24 h, arginase-1 (Figure 5A), Ym-1 (Figure 5B) and Fizz-1 (Figure 5C) were significantly induced. However, pretreatment with emodin at 10 and 50 μmol/L reduced IL-4-triggered AAM marker expression, including arginase-1, Ym-1 and Fizz-1 (Figure 5). These data suggested that emodin attenuated IL-4-induced AAM polarization.

Emodin inhibited the IL-4-mediated AAM polarization. BMDMs isolated from WT mice were cultured and pretreated with emodin at 0, 2, 10 and 50 μmol/L for 30 min and then stimulated with 10 ng/mL of IL-4 for 24 h. Total RNA was isolated, and the mRNA levels of arginase-1 (A), Ym-1 (B) and Fizz-1 (C) were quantified by quantitative real-time PCR. Statistical analysis was performed using Student's t test. n=3. * P<0.05, ** P<0.01 between the two indicated groups.
Emodin decreased IL-4-induced STAT6 activation and KLF4 expression
STAT6 is a transcription factor of IL-4 that induces AAM polarization by modulating AAM biomarkers23. Though AAM polarization is controlled by multiple different signaling pathways, IL-4 receptor-mediated STAT6 activation is a classic signal for AAM polarization. To elucidate how emodin modulates AAM polarization, we stimulated BMDMs with 10 ng/mL IL-4 for 30 min; phosphor-STAT6 (Tyr641) was induced, and emodin remarkably inhibited STAT6 activation at 10 and 50 μmol/L (Figure 6A and B). Kruppel-like factor 4 (KLF4) is a DNA-binding transcriptional regulator24, which is another vital gene for controlling AAM polarization25. Upon treatment with IL-4 for 24 h, the expression of KLF4 was increased. The increased KLF4 was inhibited by pretreatment with emodin at 10 and 50 μmol/L (Figure 6C and D). Based on the dose-dependent inhibition of STAT6 activation and KLF4 expression by emodin, we considered that emodin reduced AAM polarization via the down-regulation of STAT6 and KLF4, which in turn alleviated asthmatic inflammation.

Emodin reduced the phosphorylation of STAT6 and the expression of KLF4 in IL-4-induced BMDMs. BMDMs isolated from WT mice were pretreated with emodin at 0, 2, 10 and 50 μmol/L for 30 min and then stimulated with 10 ng/mL of IL-4. Next, Western blotting was performed to detect the expression of STAT6 and KLF4. (A) The phosphorylation of STAT6 and total STAT6 was detected at 30 min after IL-4 stimulation. (C) The expression of KLF4 and GAPDH was detected by Western blotting. The ratio of phosphorylated and total STAT6 (B) and the ratio of KLF4 and GAPDH (D) in densitometry were analyzed using ImageJ software. Statistical analysis was performed using Student's t test. n=3. * P<0.05, ** P<0.01 between the indicated groups.
Discussion
Natural substances are the most important resources in drug discovery and development processes. Emodin is a purgative resin that is widely found in Rheum palmatum, buckthorn and Polygonum multiflorum 26. In this study, we identified that emodin had a pharmacological activity in alleviating asthmatic inflammation and reducing alternatively activated macrophages via IL-4 mediated signaling. In an experimental triple allergen DRA-induced asthma model, emodin attenuated lung eosinophilic infiltration, IgE production, mucus glycol conjugates and Th2 cytokine production. In addition, our results indicated that emodin inhibited the expression of AAM markers, including arginase-1, Ym-1 and Fizz-1 both in vivo and in vitro. Upon stimulation with IL-4, emodin decreased STAT6 phosphorylation, which is a critical signal for AAM polarization. Therefore, our studies revealed emodin as a potential agent for treatment of asthmatic inflammation.
Macrophages are a type of tissue-resident innate immune cell that not only acts as a first-line defense against pathogen invasion but also modulates the tissue immune microenvironment via the production of cytokines and chemokines27,28. Recent studies have demonstrated that the diversity and plasticity of macrophages play critical roles in the modulation of severe chronic diseases, including cancer, type II diabetes, cystic fibrosis and atherosclerosis29,30. Although asthma is traditionally considered to be a type 2 immune response in which Th2 lymphocytes, B cells, mast cells and dendritic cells are of importance for pathological development, IL-4-mediated macrophage polarization also contributes to asthmatic inflammation11,29. To determine whether emodin attenuates asthmatic inflammation by modulation of the pulmonary microenvironment, we injected mice with emodin in the DRA-induced late phase of allergic responses in which adaptive immune responses have been completely established. Decreased eosinophilic inflammation indicated that emodin had effects on the later-phase allergic responses. According to the decreased expression of the AAM markers arginase-1, Ym-1 and Fizz-1 in lung tissues, emodin may ameliorate asthma by at least partly reducing AAM polarization. The role of AAMs in asthmatic lung inflammation has been proven by using MAFIA (macrophage Fas-induced apoptosis) mice. Following specific depletion of macrophages in the late-phase of allergic responses, DRA-induced asthmatic pulmonary inflammation is remarkably reduced10. Therefore, interference of AAM polarization by agents such as emodin is a potential therapeutic strategy for treating asthma.
IL-4, a type-2 cytokine, not only induces differentiation of naïve T cells to Th2 cells but also polarizes resting macrophages to AAMs31. In macrophages, IL-4 binds to IL-4 receptors (IL-4Rs) and initiates the JAK2/3/STAT6 signal pathway, leading to AAM polarization and inducing the expression of Fizz-1, arginase-1, Ym-1 and CD206. The effect of emodin on macrophage activation has been well explored. Recent studies have shown that emodin can decrease LPS-TLR4-mediated signaling pathways through inhibiting the activation of NF-κB/IRF5/STAT132 or PPARγ33. In addition, emodin also suppresses IL-1β-mediated NLRP3 inflammasome activation34 and IL-1β-induced mesangial cell proliferation and extracellular matrix production by inhibiting p38 MAPK35. Here, our results showed that emodin dose-dependently inhibited IL-4-mediated STAT6 phosphorylation, which was associated with decreased expression of Fizz-1, Ym-1 and arginase in response to IL-4 treatment31,36. Because IL-4-triggered STAT6 activation is required for AAM polarization11, emodin can suppress AAM polarization by inhibiting STAT6 phosphorylation. Given that STAT6 deficient mice are protected against experimental asthma in which STAT6 is involved in Th2 lymphocyte differentiation37, emodin may also inhibit Th2-mediated immune responses. Thus, targeting the STAT6 signal pathway is an alternative therapeutic target for the treatment of asthma. In fact, a novel STAT6 inhibitor, AS1517499 (leflunomide), attenuates ovalbumin antigen (OVA)-induced asthma38. Therefore, STAT6 inhibitory agents such as AS1517499 and emodin might be helpful for ameliorating asthma.
Emodin, a traditional Chinese medicine, has primarily been used as an anti-bacterial and anti-inflammatory agent39,40,41, whereas little is known about its effect on asthma. Previous studies have shown that emodin protects against OVA-induced asthmatic inflammation by reducing IL-4, IL-5 and MMP-9 expression and inducing HO-1 expression42. However, the underlying mechanism has not been identified. The OVA model is characterized as 'type 2-mediated immune responses', which lacks the impressive asthmatic airway remodeling or chronic changes that are found in human asthma43. Here, we used a DRA-induced asthma model instead of the traditional OVA-induced animal model to study the effect of emodin on allergic asthma. Because the triple allergens dust mite, ragweed and aspergillus are general allergens for inducing human asthma, the pathological process is more similar to that of typical clinic patients. In our study, we found that emodin exhibited protective effects against DRA-induced asthma. We also demonstrated that emodin attenuated mucus production and airway remodeling by inhibiting AAM polarization and Fizz-1 expression, not just type 2 immune responses. Therefore, we believe that emodin may be useful as a new agent to treat asthma. Recently, inhaled corticosteroids are the first-line therapy for the treatment of chronic asthma in many countries. However, several reports have shown that severe asthmatic patients cannot be adequately controlled by an ICS only. In addition, corticosteroid phobia and fears are prevalent, influencing the therapeutic efficacy and asthma control. The use of complementary and alternative medicine has become a more prevalent option for asthma patients44. For instance, previous reports have demonstrated the modulation effect of Osthole, Herba Epimedii and chrysophanol on asthmatic inflammation45,46,47. Our study determined that emodin alleviated DRA-induced asthma by interfering with AAM polarization. Different from emodin, corticosteroids have been shown to bind to glucocorticoid receptors and suppress the multiple inflammatory genes that are activated in asthmatic airways by reversing histone acetylation of the activated inflammatory genes48,49,50. Further studies should focus on the combination use of corticosteroids and emodin in the DRA-induced animal model to determine whether combined therapy could further alleviate asthma symptoms.
Our study proved that emodin had an effect on reducing asthmatic inflammation. Previous reports revealed that emodin inhibited tumor proliferation, reduced inflammation and alleviated diabetes15,19,51. Various effects of emodin indicate that it may target several genes, and future research should focus on identification of the drug targets and discovery of the mechanisms of its direct regulation. Since AAMs display different regulatory functions in various diseases except asthma, emodin may exert therapeutic effects on several chronic inflammatory diseases in addition to asthma.
Author contribution
Feng QIAN and Lei SUN designed the study and drafted the manuscript; Yun-duan SONG and Xiao-zong LI performed the experiments and drafted the manuscript; and Ya-xian WU, Yao SHEN, Fang-fang LIU, Pei-pei GAO participated in the data analysis and were involved in discussions of the experiments. All authors read and approved the final manuscript.
References
Peters SP, Ferguson G, Deniz Y, Reisner C. Uncontrolled asthma: a review of the prevalence, disease burden and options for treatment. Respir Med 2006; 100: 1139–51.
Siracusa CM, Brewington JJ, Brockbank JC, Guilbert TW. Update in pediatric lung disease 2014. Am J Respir Crit Care Med 2015; 192: 918–23.
Medoff BD, Thomas SY, Luster AD. T cell trafficking in allergic asthma: the ins and outs. Annu Rev Immunol 2008; 26: 205–32.
Levy BD, Noel PJ, Freemer MM, Cloutier MM, Georas SN, Jarjour NN, et al. Future Research Directions in Asthma. An NHLBI Working Group Report. Am J Respir Crit Care Med 2015; 192: 1366–72.
Lloyd CM, Hessel EM. Functions of T cells in asthma: more than just T(H)2 cells. Nat Rev Immunol 2010; 10: 838–48.
Kim YY, Je IG, Kim MJ, Kang BC, Choi YA, Baek MC, et al. 2-Hydroxy-3-methoxybenzoic acid attenuates mast cell-mediated allergic reaction in mice via modulation of the FcepsilonRI signaling pathway. Acta Pharmacol Sin 2017; 38: 90–9.
Na H, Cho M, Chung Y. Regulation of Th2 cell immunity by dendritic cells. Immune Netw 2016; 16: 1–12.
Xu J, Zhu YT, Wang GZ, Han D, Wu YY, Zhang DX, et al. The PPARgamma agonist, rosiglitazone, attenuates airway inflammation and remodeling via heme oxygenase-1 in murine model of asthma. Acta Pharmacol Sin 2015; 36: 171–8.
Goplen N, Karim MZ, Liang Q, Gorska MM, Rozario S, Guo L, et al. Combined sensitization of mice to extracts of dust mite, ragweed, and Aspergillus species breaks through tolerance and establishes chronic features of asthma. J Allergy Clin Immunol 2009; 123: 925–32 e11.
Lee YG, Jeong JJ, Nyenhuis S, Berdyshev E, Chung S, Ranjan R, et al. Recruited alveolar macrophages, in response to airway epithelial-derived monocyte chemoattractant protein 1/CCl2, regulate airway inflammation and remodeling in allergic asthma. Am J Respir Cell Mol Biol 2015; 52: 772–84.
Qian F, Deng J, Lee YG, Zhu J, Karpurapu M, Chung S, et al. The transcription factor PU.1 promotes alternative macrophage polarization and asthmatic airway inflammation. J Mol Cell Biol 2015; 7: 557–67.
Barnes PJ. Severe asthma: advances in current management and future therapy. J Allergy Clin Immunol 2012; 129: 48–59.
Cooper V, Metcalf L, Versnel J, Upton J, Walker S, Horne R. Patient-reported side effects, concerns and adherence to corticosteroid treatment for asthma, and comparison with physician estimates of side-effect prevalence: a UK-wide, cross-sectional study. NPJ Prim Care Respir Med 2015; 25: 15026.
Barnes PJ. New anti-inflammatory targets for chronic obstructive pulmonary disease. Nat Rev Drug Discov 2013; 12: 543–59.
Meng G, Liu Y, Lou C, Yang H. Emodin suppresses lipopolysaccharide-induced pro-inflammatory responses and NF-kappaB activation by disrupting lipid rafts in CD14-negative endothelial cells. Br J Pharmacol 2010; 161: 1628–44.
Zhong T, Zhang LY, Wang ZY, Wang Y, Song FM, Zhang YH, et al. Rheum emodin inhibits enterovirus 71 viral replication and affects the host cell cycle environment. Acta Pharmacol Sin 2017; 38: 392–401.
Liu H, Gu LB, Tu Y, Hu H, Huang YR, Sun W. Emodin ameliorates cisplatin-induced apoptosis of rat renal tubular cells in vitro by activating autophagy. Acta Pharmacol Sin 2016; 37: 235–45.
Li M, Li C, Liu WX, Liu C, Cui J, Li Q, et al. Dysfunction of PLA2G6 and CYP2C44-associated network signals imminent carcinogenesis from chronic inflammation to hepatocellular carcinoma. J Mol Cell Biol 2017.
Subramaniam A, Shanmugam MK, Ong TH, Li F, Perumal E, Chen L, et al. Emodin inhibits growth and induces apoptosis in an orthotopic hepatocellular carcinoma model by blocking activation of STAT3. Br J Pharmacol 2013; 170: 807–21.
He HQ, Wu YX, Nie YJ, Wang J, Ge M, Qian F. LYRM03, an ubenimex derivative, attenuates LPS-induced acute lung injury in mice by suppressing the TLR4 signaling pathway. Acta Pharmacol Sin 2017; 38: 342–50.
Wu YX, He HQ, Nie YJ, Ding YH, Sun L, Qian F. Protostemonine effectively attenuates lipopolysaccharide-induced acute lung injury in mice. Acta Pharmacol Sin 2018; 39: 85–96.
Lu L, Pan C, Chen L, Hu L, Wang C, Han Y, et al. AMPK activation by peri-sciatic nerve administration of ozone attenuates CCI-induced neuropathic pain in rats. J Mol Cell Biol 2017; 9: 132–43.
Zhang Y, Mei H, Chang X, Chen F, Zhu Y, Han X. Adipocyte-derived microvesicles from obese mice induce M1 macrophage phenotype through secreted miR-155. J Mol Cell Biol 2016; 8: 505–17.
Tang H, Zhu H, Wang X, Hua L, Li J, Xie Q, et al. KLF4 is a tumor suppressor in anaplastic meningioma stem-like cells and human meningiomas. J Mol Cell Biol 2017; 9: 315–24.
Atkin SL, Barrier S, Cui Z, Fletcher PD, Mackenzie G, Panel V, et al. UV and visible light screening by individual sporopollenin exines derived from Lycopodium clavatum (club moss) and Ambrosia trifida (giant ragweed). J Photochem Photobiol B Biol 2011; 102: 209–17.
Dong X, Fu J, Yin X, Cao S, Li X, Lin L, et al. Emodin: A Review of its Pharmacology, Toxicity and Pharmacokinetics. Phytother Res 2016; 30: 1207–18.
Guilliams M, Scott CL. Does niche competition determine the origin of tissue-resident macrophages? Nat Rev Immunol 2017; 17: 451–60.
Huang Q, Wang T, Wang HY. Ginsenoside Rb2 enhances the anti-inflammatory effect of omega-3 fatty acid in LPS-stimulated RAW264.7 macrophages by upregulating GPR120 expression. Acta Pharmacol Sin 2017; 38: 192–200.
Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest 2012; 122: 787–95.
Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature 2013; 496: 445–55.
Van Dyken SJ, Locksley RM. Interleukin-4- and interleukin-13-mediated alternatively activated macrophages: roles in homeostasis and disease. Ann Rev Immunol 2013; 31: 317–43.
Iwanowycz S, Wang J, Altomare D, Hui Y, Fan D. Emodin Bidirectionally Modulates Macrophage Polarization and Epigenetically Regulates Macrophage Memory. J Biol Chem 2016; 291: 11491–503.
Zhu T, Zhang W, Feng SJ, Yu HP. Emodin suppresses LPS-induced inflammation in RAW264.7 cells through a PPARgamma-dependent pathway. Int Immunopharmacol 2016; 34: 16–24.
Han JW, Shim DW, Shin WY, Heo KH, Kwak SB, Sim EJ, et al. Anti-inflammatory effect of emodin via attenuation of NLRP3 inflammasome activation. Int J Mol Sci 2015; 16: 8102–9.
Wang R, Wan Q, Zhang Y, Huang F, Yu K, Xu D, et al. Emodin suppresses interleukin-1beta induced mesangial cells proliferation and extracellular matrix production via inhibiting P38 MAPK. Life Sci 2007; 80: 2481–8.
Wynn TA. Type 2 cytokines: mechanisms and therapeutic strategies. Nat Rev Immunol 2015; 15: 271–82.
Valladao AC, Frevert CW, Koch LK, Campbell DJ, Ziegler SF. STAT6 regulates the development of eosinophilic versus neutrophilic asthma in response to alternaria alternata. J Immunol 2016; 197: 4541–51.
Chiba Y, Todoroki M, Nishida Y, Tanabe M, Misawa M. A novel STAT6 inhibitor AS1517499 ameliorates antigen-induced bronchial hypercontractility in mice. Am J Respir Cell Mol Biol 2009; 41: 516–24.
Lu J, Xu Y, Zhao Z, Ke X, Wei X, Kang J, et al. Emodin suppresses proliferation, migration and invasion in ovarian cancer cells by down regulating ILK in vitro and in vivo. Onco Targets Ther 2017; 10: 3579–89.
Li H, Yang T, Zhou H, Du J, Zhu B, Sun Z. Emodin combined with nanosilver inhibited sepsis by anti-inflammatory protection. Front Pharmacol 2016; 7: 536.
Chen GL, Zhang JJ, Kao X, Wei LW, Liu ZY. Emodin ameliorates lipopolysaccharides-induced corneal inflammation in rats. Int J Ophthalmol 2015; 8: 665–9.
Wang T, Zhong XG, Li YH, Jia X, Zhang SJ, Gao YS, et al. Protective effect of emodin against airway inflammation in the ovalbumin-induced mouse model. Chin J Integr Med 2015; 21: 431–7.
Swirski FK, Sajic D, Robbins CS, Gajewska BU, Jordana M, Stampfli MR. Chronic exposure to innocuous antigen in sensitized mice leads to suppressed airway eosinophilia that is reversed by granulocyte macrophage colony-stimulating factor. J Immunol 2002; 169: 3499–506.
Ip KI, Hon KL, Tsang YCK, Leung NHT. Steroid phobia, Chinese medicine and asthma control. Clin Respir J 2017. Doi: 10.1111/crj.12705.
Chiang CY, Lee CC, Fan CK, Huang HM, Chiang BL, Lee YL. Osthole treatment ameliorates Th2-mediated allergic asthma and exerts immunomodulatory effects on dendritic cell maturation and function. Cell Mol Immunol 2017. Doi: 10.1038/cmi.2017.71
Tang X, Nian H, Li X, Yang Y, Wang X, Xu L, et al. Effects of the combined extracts of Herba Epimedii and Fructus Ligustrilucidi on airway remodeling in the asthmatic rats with the treatment of budesonide. BMC Complement Altern Med 2017; 17: 380.
Witz L, Josefsberg Z, Kaufman H, Laron Z. When should hydrocortisone therapy be instituted in children with hypopituitarism? Am J Dis Child 1988; 142: 881–3.
Barnes PJ, Pedersen S. Efficacy and safety of inhaled corticosteroids in asthma. Report of a workshop held in Eze, France, October 1992. Am Rev Respir Dis 1993; 148: S1–26.
Barnes PJ, Adcock IM. How do corticosteroids work in asthma? Ann Intern Med 2003; 139: 359–70.
Trevor JL, Deshane JS. Refractory asthma: mechanisms, targets, and therapy. Allergy 2014; 69: 817–27.
Feng Y, Huang SL, Dou W, Zhang S, Chen JH, Shen Y, et al. Emodin, a natural product, selectively inhibits 11beta-hydroxysteroid dehydrogenase type 1 and ameliorates metabolic disorder in diet-induced obese mice. Br J Pharmacol 2010; 161: 113–26.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (No 81373424, 81573438, 81773741 and 81770633) and the Shanghai Pudong Hospital Talent Promotion Project (No PJ201601).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Song, Yd., Li, Xz., Wu, Yx. et al. Emodin alleviates alternatively activated macrophage and asthmatic airway inflammation in a murine asthma model. Acta Pharmacol Sin 39, 1317–1325 (2018). https://doi.org/10.1038/aps.2017.147
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2017.147
Keywords
This article is cited by
-
Repositioning baloxavir marboxil as VISTA agonist that ameliorates experimental asthma
Cell Biology and Toxicology (2024)
-
Emodin Alleviates the Th17/Treg Imbalance by Targeting Myd88/NF-κB Signaling in Subacute Thyroiditis
Revista Brasileira de Farmacognosia (2023)
-
Rhubarb Alleviates Acute Lung Injury by Modulating Gut Microbiota Dysbiosis in Mice
Current Microbiology (2022)
-
Advances in the study of emodin: an update on pharmacological properties and mechanistic basis
Chinese Medicine (2021)
-
Emodin alleviated pulmonary inflammation in rats with LPS-induced acute lung injury through inhibiting the mTOR/HIF-1α/VEGF signaling pathway
Inflammation Research (2020)
