
Abstract
Aim:
Deacetylisovaltratum (DI) is isolated from the traditional Chinese herbal medicine Patrinia heterophylla Bunge, which exhibits anti-cancer activity. Here, we investigated the effects of DI on human gastric carcinoma cell lines in vitro and elucidated its anti-cancer mechanisms.
Methods:
Human gastric carcinoma AGS and HGC-27 cell lines were treated with DI, and cell viability was detected with MTT assay. Cell cycle stages, apoptosis and mitochondrial membrane potential were measured using flow cytometry. Protein levels were analyzed by Western blotting. Tubulin polymerization assays and immunofluorescence were used to characterize the tubulin polymerization process.
Results:
DI inhibited the cell viability of AGS and HGC-27 cells in a dose- and time-dependent manner with IC50 values of 12.0 and 28.8 μmol/L, respectively, at 24 h of treatment. Treatment with DI (10–100 μmol/L) dose-dependently promoted tubulin polymerization, and induced significant G2/M cell cycle arrest in AGS and HGC-27 cells. Moreover, DI treatment disrupted mitochondrial membrane potential and induced caspase-dependent apoptosis in AGS and HGC-27 cells.
Conclusion:
DI induces G2/M-phase arrest by disrupting tubulin polymerization in human gastric cancer cells, which highlights its potent anti-cancer activity and potential application in gastric cancer therapy.
Similar content being viewed by others


Introduction
Gastric cancer (GC) is the fourth most common cancer and the second leading cause of cancer-related death worldwide1. GC is often diagnosed at an advanced stage, and the prognosis remains very poor, with an average 5-year overall survival rate of 20%2,3. Despite tremendous efforts to optimize therapeutic strategies for treating gastric cancer, the standard chemotherapy regimen is still controversial in gastric cancer due to the complexity of GC staging4. Therefore, exploring efficacious compounds is a prevailing issue in gastric cancer research that scientists will urgently need to address in the near future.
The traditional Chinese herbal medicine Patrinia heterophylla Bunge has been used for centuries to treat metrocarcinoma and cervical cancer. However, except for recently published studies reporting potentially active compounds, there is scant information on the bioactive components of this species5,6,7,8. Furthermore, the underlying mechanisms of its anti-cancer activity remain largely unknown. Deacetylisovaltratum (DI) is a novel compound isolated from P heterophylla Bunge with good purity (≥98.0%) based on preparative thin layer chromatography (TLC) and high-performance liquid chromatography (HPLC), and its structure was determined by H-NMR7,9.
In the present study, we found that DI effectively caused G2/M-phase arrest in gastric cancer cells by disrupting tubulin polymerization. In addition, prolonged treatment of DI induced mitochondrial and caspase-dependent apoptosis. Therefore, DI shows promise as a potent anti-cancer agent. Determination of the molecular target of DI will shed further light on the exploration of natural compounds effective against gastric cancer.
Materials and methods
Cell culture
F12, RPMI-1640 medium and fetal bovine serum (FBS) were purchased from Gibco, BRL (Grand Island, NY, USA). The Cycletest Plus DNA Reagent Kit was purchased from BD Biosciences (Franklin Lakes, NY, USA). Hoechst33258 was obtained from Sigma-Aldrich (St Louis, MO, USA). The Annexin V-FITC Apoptosis Kit was purchased from BestBio (Shanghai, China). The Mitochondrial Membrane Potential Assay Kit was procured from Signalway Antibody (College Park, MD, USA). The Tubulin Polymerization Assay Kit was purchased from Cytoskeleton Inc (Denver, CO, USA). Primary antibodies were purchased from Abcam Inc (Cambridge, MA, USA). Human gastric carcinoma AGS and HGC-27 cell lines were purchased from the Chinese Academy of Sciences (Shanghai, China). Cells were cultured in F12 or RPMI-1640 medium containing 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin at 37 °C in a 5% CO2 humidified atmosphere. Deacetylisovaltratum (DI) was dissolved in DMSO at a concentration of 100 mmol/L.
Cell viability assay
Cell proliferation was measured by the MTT assay. Cells (3×103/well) were cultured in 96-well plates for 24 h and treated with various concentrations (2.5, 5, 10, 15, 20, 30, and 40 μmol/L) of DI. After 24, 48, and 72 h treatment, 50 μL of MTT solution (5 mg/mL in PBS) was added to each well, and the cells were cultured for another 4 h at 37 °C. The supernatant was completely removed, and formazan was solubilized with 100 μL DMSO. Cell viability was quantified at 570 nm using a Multiskan Spectrum spectrophotometer (Thermo Scientific, Rockford, IL, USA).
Colony formation assay
AGS and HGC-27 cells were seeded in 6-well plates at the density of 1×103/well and incubated for 72 h. The cells were then treated with various concentrations (4, 8, 10, and 20 μmol/L) of DI. After 7 d, the cells were fixed with 4% paraformaldehyde for 15 min and stained with Giemsa solution for another 15 min. Visible colonies were photographed using the ChemiDoc XPS system (Bio-Rad, Hercules, CA, USA).
Cell cycle analysis
After treatment with DI, AGS, and HGC-27 cells were harvested and washed twice with cold PBS and then fixed in 70% cold ethanol at 4 °C overnight. The cells were stained using the Cycletest Plus DNA Reagent Kit according to the manufacturer's instructions (BD Bioscience, San Jose, CA, USA). Cell cycle distribution was analyzed using a flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA).
Detection of mitochondrial membrane potential
Mitochondrial membrane potential was visualized by staining with 5,5′,6,6′-tetrachloro-1,1′,3,3′ tetraethyl-imidacarbocyanine iodide (JC-1). Cells were seeded in 6-well plates at the density of 2×105/well and cultured for 24 h. After treatment, the cells were collected, washed with PBS, and incubated with JC-1 for 15 min at 37 °C. After removing the dye by washing, the cells were immediately analyzed using a flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA).
Hoechst 33342 staining
AGS and HGC-27 cells (2×104 cells/well) were cultured in 24-well plates. After exposure to DI, the cells were fixed with 4% paraformaldehyde for 20 min and stained with Hoechst 33342 for 20 min at 37 °C. After washing with PBS, the cells were observed under a fluorescence microscope (Nikon, Ti-E, Japan).
Apoptosis assay
Treated or untreated cells were harvested and washed with PBS. The cells were then stained with the Annexin V-FITC Apoptosis Kit according to the manufacturer's instructions and analyzed by flow cytometry (Becton Dickinson, Franklin Lakes, NJ, USA).
Western blot analysis
After treatment with different concentrations of DI, total protein was extracted using RIPA Lysing Buffer as previously described10. The protein sample (40 μg) was subjected to 12% SDS-PAGE and transferred to a PVDF membrane (Bio-Rad, Hercules, CA, USA). The membranes were blocked with 5% non-fat milk at room temperature for 1 h and then incubated with specific primary antibodies overnight at 4 °C. After washing with TBST, the membranes were incubated with secondary antibodies at room temperature for another 1 h. The protein bands were visualized using the ECL system WBKLS0050 (EMD Millipore, Billerica, MA, USA) and analyzed using Bio-Rad Laboratories Quantity One software (Bio-Rad, Hercules, CA, USA).
In vitro tubulin polymerization assay
The tubulin polymerization assay was performed as described by the manufacturer (Cytoskeleton Inc, Denver, CO, USA). Briefly, DI at various concentrations was incubated with purified bovine tubulin in 80 mmol/L PIPES buffer (pH 6.9) containing 2 mmol/L MgCl2, 0.5 mmol/L EGTA, 1 mmol/L GTP and 15% glycerol. Increases in absorbance at 340 nm were recorded every min for 1 hat 37 °C using a spectrophotometer.
Immunofluorescence
AGS and HGC-27 cells were plated incomplete medium in SigmaNunc®Lab-Tek®IIChamber Slide systems (8 chamber; Sigma-Aldrich, St Louis, MO, USA) at 10 000 cells per chamber and incubated for 24 h before use. The medium was replaced with DI (12 or 30 μmol/L) or paclitaxel (0.5 μmol/L)11 and cultured for 24 h. The cells were then rinsed with PBS twice before fixation by 4% formaldehyde for 20 min at room temperature. The cells were rinsed with PBS three times and permeabilized with 0.2% Triton X-100 for 10 min. After washing with PBS, the cells were blocked with 5% BSA for 30 min and incubated overnight with the primary antibody at 4 °C. The cells were washed with PBS and incubated with the secondary antibody (Goat Anti-Rabbit IgG H&L, Alexa Fluor® 594, Abcam, Cambridge, MA, USA) for 30 min. The slides were sealed with cover glasses using the ProLong® Gold anti-fade reagent with DAPI (Invitrogen™, Thermo Fisher Scientific, Waltham, MA, USA) and immediately observed by confocal microscopy (Leica SP8, Mannheim, Germany). Tubulin was visualized at 552 nm excitation, and fluorescence emission was observed using a 570–710 nm bandpass filter. DAPI was excited at 405 nm, and emission was detected from 420 to 540 nm.
Statistical analysis
The results are expressed as the mean±SD of at least three independent experiments. Differences between mean values were analyzed using Student's t-test and were considered statistically significant when P<0.05. Graphs were prepared using SigmaPlot 12.0 software.
Results
DI inhibited the proliferation of AGS and HGC-27 cells
After treatment with DI, both AGS and HGC-27 cells showed markedly reduced viability in a dose- and time-dependent manner (Figure 1A). Treatment of AGS cells with 10 μmol/L DI resulted in approximately 40% of survival fraction compared with untreated cells. In the same experimental panel, HGC-27 cells were slightly less sensitive to DI than AGS cells. The IC50 values were 12.0 and 28.8 μmol/L for AGS and HGC-27 cells, respectively, at 24 h of treatment. In addition, 4 μmol/L DI caused notable inhibition of colony formation by AGS cells (Figure 1B). However, apparent colony inhibition in HGC-27 cells was achieved with 10 μmol/L of DI, which was consistent with the cytotoxicity results that HGC-27 cells were less sensitive than AGS cells to DI.

DI inhibited the proliferation of AGS and HGC-27 cells. DI inhibited the proliferation of AGS and HGC-27 cells in a dose- and time-dependent manner (A). The indicated concentrations of DI notably inhibited colony formation by both AGS and HGC-27 cells (B). Cells were treated with DI for 24 h and cultured for another 7 d before Giemsa staining.
DI caused G2/M-phase arrest in AGS and HGC-27 cells
Cells were treated with DI for various times and at various concentrations and then analyzed by flow cytometry. As shown in Figure 2, 12 h of treatment with 12 μmol/L DI resulted in an AGS cell population in G2/M phase that was twice that of the untreated cells. For HGC-27 cells, the cell proportion in G2/M phase gradually increased along with treatment time, reaching the highest proportion at 12 h (1.5-fold increase compared with untreated cells).

DI caused cell cycle arrest in AGS (A) and HGC-27 (B) cells. AGS and HGC-27 cells were treated with 12 and 30 μmol/L DI, respectively, for 6 and 12 h. The cells were then stained with PI and analyzed by flow cytometry. Each bar represents the mean±SD. *P<0.05, **P<0.01.
DI interfered with tubulin polymerization dynamics
The G2/M-phase transition is controlled by a complex consisting of CyclinB and cdc2; this complex is further regulated by cdc25c, and Chk1 is an upstream regulator of cdc25c. After treatment with DI, expression of cdc2 and cdc25c proteins was not greatly altered with treatment duration (Figure 3A), showing that the Chk1-cdc25c-cdc2 signaling pathway might not play a critical role in DI-induced G2/M arrest.

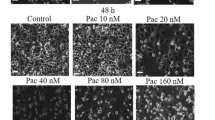
Effects of DI on cell cycle regulatory proteins and tubulin polymerization. AGS (left panel) and HGC-27 (right panel) cells were treated with 12 and 30 μmol/L DI, respectively, for indicated time, and Western blot analysis was performed (A). Polymerization of microtubules in the presence of various concentrations of DI or paclitaxel was recorded continuously for 60 min by measuring absorbance at 340nm (B). Immunofluorescence was used to probe intracellular tubulin (C). Cells were treated with DI or paclitaxel for 12 h and then fixed, permeabilized and incubated with primary and secondary antibodies. Tubulin was visualized by excitation at 552 nm; fluorescence emission was observed using a 570–710 nm bandpass filter. DAPI was excited at 405 nm, and emission was detected from 420 to 540 nm.
A tubulin dynamics assay in a cell-free system was used to investigate the effect of DI on tubulin polymerization (Figure 3B). Drug-free tubulin polymerized gradually under physiological conditions. Paclitaxel, which is known to accelerate the velocity of tubulin polymerization, was used as a positive control. In this study, the polymerization curve of paclitaxel-treated tubulin exhibited a sharp upward slope compared with that of drug-free tubulin or DI-treated tubulin. Although low concentrations of DI resulted in only a mild upward slope, as illustrated in Figure 3B, DI at 100 μmol/L induced tubulin polymerization to a much higher degree.
To further confirm these results in an intracellular system, immunofluorescence was used to probe intracellular tubulin before and after treatment. DI at 12 and 30 μmol/L caused intense fluorescence in AGS and HGC-27 cells, respectively, compared with untreated cells. Moreover, distinct differences in the organization of microtubule arrays were observed between untreated and DI-treated cells, which was similar to paclitaxel-treated cells (Figure 3C). Therefore, DI could share paclitaxel's ability to promote tubulin polymerization.
DI induced loss of mitochondrial membrane potential
Measurement of mitochondrial membrane potential is one of the methods used to detect changes in mitochondrial membrane polarization and apoptosis initiation. To explore the effect of DI on mitochondrial membrane potential, cells were cultured in the presence of DI for 24 h. With increasing DI concentration, the fluorescence generated from JC-1-stained cells gradually shifted from red to green, suggesting a decrease in mitochondria membrane potential (Figure 4A and 4B). Therefore, DI disrupted mitochondrial membrane potential in a dose-dependent manner in both AGS and HGC-27 cells.

DI disrupted mitochondrial transmembrane permeability. AGS (A) and HGC-27 (B) cells were treated with different concentrations of DI for 24 h and stained with JC-1 dye, followed by flow cytometry analysis. DI caused apoptosis in AGS and HGC-27 cells. AGS (C) and HGC-27 (D) cells before and after treatment with DI for 24 h were stained with Annexin V-FITC/PI, followed by flow cytometry analysis. Cellular morphological changes were examined by Hoechst 33342 staining and observed under fluorescence microscopy (E). Each bar represents the mean±SD. **P<0.01.
DI induced apoptosis in AGS and HGC-27 cells
Flow cytometry was employed to analyze early and late apoptotic cells before and after DI treatment. For AGS cells, the total number of apoptotic cells (including Annexin V+ and PI+) increased significantly (4-fold compared as control) after treatment with 16 μmol/L of DI (Figure 4C). For HGC-27 cells, 30 μmol/L DI resulted in a 5-fold increase in the total number of apoptotic cells (Figure 4D).
To further assess DI-induced apoptosis, DI-treated cells were stained with Hoechst 33342 and observed under fluorescence microscopy (Figure 4E). Compared to the uniformly distributed blue fluorescence in untreated cells, the cells treated with increasing concentrations of DI exhibited shrunken size, bright staining, chromatin condensation or fragmented nuclei. More importantly, apoptotic bodies were observed at high DI concentrations (yellow arrows in Figure 4E).
The effect of DI on proteins regulating cell cycle and apoptosis
In both AGS and HGC-27 cells, active caspase-3 gradually increased in response to high concentrations of DI, which further contributed to PARP cleavage. In addition, procaspase-9 was found to decrease after DI treatment, indicating that the mitochondrion-caspase pathway might be involved in the observed DI-induced apoptosis (Figure 5A). As 12 h of DI treatment induced G2/M-phase arrest, DNA damage was examined after the installation of DNA replication. The appearance of γ-H2AX at 24 h after exposure to the highest concentration of DI indicated apoptotic DNA fragmentation.

The effect of DI on apoptotic and cell proliferative proteins. (A) AGS and HGC-27 cells were treated with the indicated concentrations of DI for 24 h before Western blot assay. (B) Proteins regulating cell proliferation were determined after treatment with DI.
DI significantly reduced the level of p-Stat 3 after an extremely short treatment time, an event that was further found to be independent of JAK 2. Furthermore, the DNA damage predictors p-ATM and p-ATR were unchanged after DI treatment, suggesting that DI-induced G2/M arrest directly caused mitochondrial and caspase-dependent apoptosis.
Discussion
The reported anticancer effect of Patrinia heterophylla Bunge has been barely found6,7. DI is a novel compound with a specific structure extracted from this plant. Our study showed that DI possesses an anti-tumor activity by inducing cell cycle arrest and apoptosis.
It is well known that cyclin proteins and cyclin-dependent kinase are two key components of cell cycle regulation12. The G2/M checkpoint is controlled by cdc2 and CyclinB, and the activated CyclinB/cdc2 complex is important for the transition from G2 to M phase13,14. After formation of the CyclinB/cdc2 complex, cdc25c allows progression to mitosis15,16. In our study, both AGS and HGC-27 cells exhibited obvious G2/M-phase arrest by flow cytometric analysis after treatment with DI for 12 h. However, regulatory proteins including p-Chk1, p-cdc25c and p-cdc2 were not altered with DI treatment. These results caused us to speculate that DI treatment would evoke M-phase arrest, rather than G2-phase arrest, in gastric cancer cells. Accordingly, we then examined whether DI could disrupt tubulin polymerization, a function exhibited by paclitaxel17,18. Interestingly, DI accelerated tubulin polymerization in a dose-dependent manner compared with the untreated group in a cell-free system (Figure 3B). We further validated the effect of DI by probing intracellular tubulin using immunofluorescence (Figure 3C) and found that DI shares with paclitaxel the ability to stabilize polymerized tubulin in both AGS and HGC-27 cells, as suggested by intensified red fluorescence in DI- and paclitaxel-treated cells (Figure 3C).
Prolonged treatment with DI generally causes gradual DNA damage, resulting in ATR phosphorylating and activating Chk1, which in turn reduces the level of cdc25c protein19,20. In our study, neither p-ATM nor p-ATR was altered with treatment duration (Figure 5B). In contrast, p21, which is strongly induced upon DNA damage and plays a pivotal role in the G2/M checkpoint21,22,23, was dramatically up-regulated after treatment with DI for 6 h (Figure 3A). These results suggest a possible role for p21 in DI-induced cell cycle arrest.
The caspase family is a group of critical proteins regulating apoptosis; the members of this family participate in the proteolytic pathways required for executing programmed cell death or apoptosis6,24. In particular, caspase-3 is one of the key mediators of apoptosis and is responsible for the proteolytic cleavage of many key proteins, such as PARP25,26. Active caspase-3 and cleaved PARP were found after DI treatment in our study. In addition, a depolarized mitochondrial membrane potential was detected using JC-1 staining. Thus, our data suggest that DI induced mitochondrial and caspase-dependent apoptosis with prolonged treatment. Lastly, an extremely short DI treatment time (0.5 h) greatly reduced expression of p-Stat 3, which was found to be JAK2 independent. However, as additional analysis using siRNA to knockdown Stat 3 expression did not undermine the effect of DI (data not shown), future work is needed to identify the molecular target of DI.
To summarize, our study demonstrated that DI caused G2/M-phase arrest by interfering with the tubulin polymerization process in both AGS and HGC-27 cells and prolonging the DI treatment time could further induce mitochondrial and caspase-dependent apoptosis. Therefore, full exploration of DI may help to elucidate the molecular mechanisms of the active constituents of P heterophylla Bunge and provide alternative therapeutic regimens for the treatment of human gastric cancer.
Author contribution
Dan ZHANG, Bo ZHANG, and Neng-ming LIN designed the research; Bo YANG contributed DI; Dan ZHANG, Li-xin ZHOU, Jun ZHAO, Yang-ling LI, and Jian-mei ZENG performed the research; You-you YAN and Lin-ling WANG analyzed the data; Dan ZHANG, Bo ZHANG, and Neng-ming LIN wrote the manuscript.
References
Fock KM . The epidemiology and prevention of gastric cancer. Aliment Pharmacol Ther 2014; 40: 250–60.
Siegel R, Naishadham D, Jemal A . Cancer statistics, 2013. CA Cancer J Clin 2013; 63: 11–30.
Tan IB, Ivanova T, Lim KH, Ong CW, Deng N, Lee J, et al. Intrinsic subtypes of gastric cancer, based on gene expression pattern, predict survival and respond differently to chemotherapy. Gastroenterology 2011; 141: 476–85.
Scartozzi M, Galizia E, Verdecchia L, Berardi R, Antognoli S, Chiorrini S, et al. Chemotherapy for advanced gastric cancer: across the years for a standard of care. Expert Opin Pharmacother 2007; 8: 797–808.
Yang B, Wang YQ, Cheng RB, Chen JL, Chen J, Jia LT, et al. Induction of cytotoxicity and apoptosis in human gastric cancer cell SGC-7901 by isovaltrate acetoxyhydrin isolated from Patrinia heterophylla Bunge involves a mitochondrial pathway and G2/M phase cell cycle arrest. Asian Pac J Cancer Prev 2013; 14: 6481–6.
Yang B, Li N, Wang YQ, Chen J, Zhang RS . Preliminary evaluation of antitumor effect and induction apoptosis in PC-3 cells of extract from Patrinia heterophylla. Rev Bras Pharmacog 2011; 21: 471–6.
Lu WZ, Li QW, Li J, Liu FZ, Yang XL . Polysaccharide from Patrinia heterophylla Bunge inhibits HeLa cell proliferation through induction of apoptosis and cell cycle arrest. Labmedicine 2009; 40: 161–6.
Lu WZ, Geng GX, Li QW, Li J, Liu FZ, Han ZS . Antitumor activity of polysaccharides isolated from Patrinia heterophylla. Pharm Biol 2010; 48: 1012–7.
Finner E, David S, Thies PW . On the active agents of Valerian. Planta Med 1984; 50: 4–6.
Nam HJ, Ching KA, Kan J, Kim HP, Han SW, Im SA, et al. Evaluation of the antitumor effects and mechanisms of PF00299804, a Pan-HER inhibitor, alone or in combination with chemotherapy or targeted agents in gastric cancer. Mol Cancer Ther 2012; 11: 439–51.
Peng T, Wu JR, Tong LJ, Li MY, Chen F, Leng YX, et al. Identification of DW532 as a novel anti-tumor agent targeting both kinases and tubulin. Acta Pharmacol Sin 2014; 35: 916–28.
Sanchez-Martinez C, Gelbert LM, Lallena MJ, de Dios A . Cyclin dependent kinase (CDK) inhibitors as anticancer drugs. Bioorg Med Chem Lett 2015; 25: 3420–35.
Liu WT, Chen C, Lu IC, Kuo SC, Lee KH, Chen TL, et al. MJ-66 induces malignant glioma cells G2/M phase arrest and mitotic catastrophe through regulation of cyclin B1/Cdk1 complex. Neuropharmacology 2014; 86: 219–27.
Wang Z, Fan M, Candas D, Zhang TQ, Qin L, Eldridge A, et al. Cyclin B1/Cdk1 coordinates mitochondrial respiration for cell-cycle G2/M progression. Dev Cell 2014; 29: 217–32.
Yu Y, Wang XY, Sun L, Wang YL, Wan YF, Li XQ, et al. Inhibition of KIF22 suppresses cancer cell proliferation by delaying mitotic exit through upregulating CDC25C expression. Carcinogenesis 2014; 35: 1416–25.
Kuo KL, Lin WC, Ho IL, Chang HC, Lee PY, Chung YT, et al. 2-Methoxyestradiol induces mitotic arrest, apoptosis, and synergistic cytotoxicity with arsenic trioxide in human urothelial carcinoma cells. PLoS One 2013; 8: e68703.
Kaur R, Kaur G, Gill RK, Soni R, Bariwal J . Recent developments in tubulin polymerization inhibitors: an overview. Eur J Med Chem 2014; 87: 89–124.
Thomas GE, Sreeja JS, Gireesh KK, Gupta H, Manna TK . +TIP EB1 downregulates paclitaxelinduced proliferation inhibition and apoptosis in breast cancer cells through inhibition of paclitaxel binding on microtubules. Int J Oncol 2015; 46: 133–46.
Liu Y, Gao F, Jiang H, Niu L, Bi Y, Young CY, et al. Induction of DNA damage and ATF3 by retigeric acid B, a novel topoisomerase II inhibitor, promotes apoptosis in prostate cancer cells. Cancer Lett 2013; 337: 66–76.
Zhao Y, Wu Z, Zhang Y, Zhu L . HY-1 induces G2/M cell cycle arrest in human colon cancer cells through the ATR-Chk1-Cdc25C and Weel pathways. Cancer Sci 2013; 104: 1062–6.
Dutto I, Tillhon M, Cazzalini O, Stivala LA, Prosperi E . Biology of the cell cycle inhibitor p21(CDKN1A): molecular mechanisms and relevance in chemical toxicology. Arch Toxicol 2015; 89: 155–78.
Borodkina AV, Shatrova AN, Deryabin PI, Grukova AA, Nikolsky NN, Burova EB . Tetraploidization or autophagy: the ultimate fate of senescent human endometrial stem cells under ATM or p53 inhibition. Cell Cycle 2016; 15: 117–27.
He PX, Zhang J, Che YS, He QJ, Chen Y, Ding J . G226, a new epipolythiodioxopiperazine derivative, triggers DNA damage and apoptosis in human cancer cells in vitro via ROS generation. Acta Pharmacol Sin 2014; 35: 1546–55.
Wang L, Hu T, Shen J, Zhang L, Chan RL, Lu L, et al. Dihydrotanshinone I induced apoptosis and autophagy through caspase dependent pathway in colon cancer. Phytomedicine 2015; 22: 1079–87.
Errami Y, Naura AS, Kim H, Ju J, Suzuki Y, El-Bahrawy AH, et al. Apoptotic DNA fragmentation may be a cooperative activity between caspase-activated deoxyribonuclease and the poly(ADP-ribose) polymerase-regulated DNAS1L3, an endoplasmic reticulum-localized endonuclease that translocates to the nucleus during apoptosis. J Biol Chem 2013; 288: 3460–8.
Sui CG, Meng FD, Li Y, Jiang YH . Antiproliferative activity of rosamultic acid is associated with induction of apoptosis, cell cycle arrest, inhibition of cell migration and caspase activation in human gastric cancer (SGC-7901) cells. Phytomedicine 2015; 22: 796–806.
Acknowledgements
This work was supported by the Zhejiang Traditional Chinese Medicine Science and Technology Project (2014ZB078, Li-xin ZHOU), Hangzhou Science and Technology Project (20140733Q04, Li-xin ZHOU), Zhejiang Provincial Program for the Cultivation of High-level Innovative Health Talents (2010-190-4, Neng-ming LIN), Zhejiang Science and Technology Project (2014C33215, Bo YANG), Natural Science Foundation of Zhejiang Province (LQ15H310001, Bo ZHANG), and Scientific Research Foundation of Zhejiang Health and Family Planning Commission (2015, KYA177, Bo ZHANG).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Zhang, D., Zhang, B., Zhou, Lx. et al. Deacetylisovaltratum disrupts microtubule dynamics and causes G2/M-phase arrest in human gastric cancer cells in vitro. Acta Pharmacol Sin 37, 1597–1605 (2016). https://doi.org/10.1038/aps.2016.91
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2016.91
