
Abstract
Glioma accounts for the majority of human brain tumors. With prevailing treatment regimens, the patients have poor survival rates. In spite of current development in mainstream glioma therapy, a cure for glioma appears to be out of reach. The infiltrative nature of glioma and acquired resistance substancially restrict the therapeutic options. Better elucidation of the complicated pathobiology of glioma and proteogenomic characterization might eventually open novel avenues for the design of more sophisticated and effective combination regimens. This could be accomplished by individually tailoring progressive neuroimaging techniques, terminating DNA synthesis with prodrug-activating genes, silencing gliomagenesis genes (gene therapy), targeting miRNA oncogenic activity (miRNA-mRNA interaction), combining Hedgehog-Gli/Akt inhibitors with stem cell therapy, employing tumor lysates as antigen sources for efficient depletion of tumor-specific cancer stem cells by cytotoxic T lymphocytes (dendritic cell vaccination), adoptive transfer of chimeric antigen receptor-modified T cells, and combining immune checkpoint inhibitors with conventional therapeutic modalities. Thus, the present review captures the latest trends associated with the molecular mechanisms involved in glial tumorigenesis as well as the limitations of surgery, radiation and chemotherapy. In this article we also critically discuss the next generation molecular therapeutic strategies and their mechanisms for the successful treatment of glioma.
Similar content being viewed by others
Introduction
Central nervous system (CNS) tumors are considered to be the most devastating of all cancers since they predominantly affect the cells of the brain or spinal cord that are most vital in regulating neurological balance1. These tumors affect people of all ages due to either developmental abnormalities or inheritance (www.cdc.gov). Age-sex-race-specific prevalence data from the 2013 CBTRUS assessed 69 720 new cases of brain tumors. The different cells involved in CNS tumors are glial cells, non-neuronal cells and Schwann cells. The classification of CNS tumors is depicted in Figure 1. Seventy percent of brain cancer and one-fifth of spinal cord cancer are glial-cell-specific2. The risk factors involved in glial tumorigenesis are exposure to chemicals, ionizing radiation, viral infection and genetic manipulation (TP53, PTEN, CDKN2A, EGFR, TSC, IDH, histone, and FGFR-TACC, etc). Glioma is characterized by high proliferative potential, infiltrative growth behavior, intratumoral heterogeneity and tumor recurrence. The location and size of glial tumors are the analytical factors that contribute to the monitoring and implementation of an appropriate treatment regimen. The mainstream treatment modality for glioma revolves around surgery, radiation and chemotherapy. These strategies are inadequate in comparison with the varied avenues of glioma progression. Surgical resection is futile due to regrowth of tumors, acute morbidity and the need for ventriculoperitoneal shunting. Radiation therapy is palliative because of normal tissue toxicity and resistance. Radiation oncologists hesitate to re-treat local recurrences, assuming loss in neuroregeneration potential. Chemotherapy is now a standard of care following surgery along with radiotherapy. Depending on the roles of different growth factors (PDGF, EGF, IGF, FGF, CNTF, VEGF, and TGF, etc) in brain tumor development, a chemotherapeutic regimen can be designed. However, bypassing the blood-brain barrier (BBB), interaction with anti-seizure medications and/or steroids, and intrinsic or acquired resistance are the limiting factors for chemotherapy.

Classification of CNS tumors. Molecular & genetic anomalies and involvement of growth factors in gliomagenesis. The CNS tumors are categorized on the basis of type of cells present in CNS and glioma is further classified on basis of type of glial cells present. The neural stem cells differentiate into different cell lineages of the CNS and putative cells of origin of glioma. Three main types of cells in the mature CNS, including neurons and glial cells (particularly oligodendrocytes and astrocytes; ependymal cells) originates during the differentiation process. The glioma originates from the direct transformation of neural stem cells or glial progenitor cells. Glial tumorigenesis is driven by upregulation or downregulation of various growth factor receptor signaling pathways. Several growth factor receptors, such vascular endhotelial growth factor receptor (VEGFR), platelet-derived growth factor receptor (PDRGF), epidermal growth factor receptor (EGFR), cyclin-dependent kinase 4 (CDK4), phosphoinositide 3-kinase (PI3K), isocitrate dehydrogenase 1(IDH1) and other growth factors receptors are overexpressed, amplified and/or mutated in gliomas. It also comprises of loss of tumor suppressor genes TP53, the retinoblastoma (Rb) gene, which are essential for cell growth, differentiation and function. Loss of heterozygosity (LOH) is most frequent genetic alteration in both primary and secondary GBMs.
This review summarizes existing treatment regimens (surgical, radiation therapy and chemotherapy) for glioma and their prevailing limitations, and addresses a vision towards the development of new glioma therapeutics, including unconventional treatment strategies such as proteogenomic characterization, identification of molecular targets initiating metastasis, and gene/microRNAs (miRNA)/stem cell/immune therapy to curb glioma. The proteogenomic characterization of glioma implicates a positive correlation among genotype, proteotype and clinical phenotype, facilitating biomarker discovery, diagnosis and design of potential therapeutics. In gene therapy, using RNAi and siRNA delivery gliomagenesis genes are silenced and DNA synthesis is terminated by prodrug-activating genes. miRNAs can be targeted because their up-regulation shows activation of both oncogenes and tumor suppressor genes. Cancer stem cells (CSCs) of the brain imitate the neural stem cell niche. The molecular characteristics exhibited by CSCs include the expression of multidrug-resistance genes (such as ABCG2 and BCRP1) and the promotion of drug efflux and CSC survival. Thus, selective annihilation of CSCs could be achieved through a combination of chemotherapy and RT with antiangiogenic drugs. Researchers hope that vaccines currently in clinical trials can effectively address the issue of tolerance so that cancer cells can be recognized by a patient's immune system. Fascinating results have been observed in patients with malignant glioma, anaplastic astrocytoma and glioma who have been vaccinated using tumor lysate as an antigen source. An efficient depletion of tumor-specific CSCs has been observed with cytotoxic T lymphocytes (CTLs) generated by dendritic cell (DC) vaccination+CSC-derived tumor lysate. Studies of the SOX2 gene have led to the genesis of specific CTLs. These strategies will encourage effective glioma stratification.
Types of glioma and their histological features
A glioma is a tumor of the glial cells that maintain the brain and nourish nerve cells. Glioma accounts for 30% of brain cancer, and 80% of gliomas are malignant. The severity of a brain tumor is due to its infiltrative nature (www.dana.org). The WHO grading system relies on atypia, mitosis, endothelial proliferation and necrosis. Tumors with none of these features are grade I and with any of these features are grade II. Grade I and II tumors are considered benign; grade III are malignant and grade IV (glioblastoma) are the most aggressive and malignant. Low-grade astrocytoma is commonly found in children, and high-grade is more frequently found in adults. The classification of glioma on the basis of the type of glial cells and their specific histological features has been discussed in Table 1. Such a glioma classification based on a broad-scale omics study would be an attempt to obtain an integrative view of glioma biology. Knowledge obtained at the system level would aid in deciphering biological insights into the molecular mechanisms underlying the limitations in prevailing glioma therapy and radio/chemo resistance, biomarker discovery, diagnosis and the design of potential therapeutics. The common approaches employed in glioma proteomics are tissue preparation, protein/peptide enrichment and separation, mass spectrometry, quantification and data analysis10,11. The insights provided by an omics study in search of protein signatures and biomarkers for glioma, highlighting the expression of specific proteins in different grades of glioma, have been tabulated in Table 2.
Low-grade glioma is most common in children, whereas in adults, diffuse high-grade gliomas (HGGs) are predominant. In children, diffuse HGGs are rare but have the same dismal prognosis as in adults and, in terms of histopathology, clinical behavior, genetic expression signatures and genetic abnormalities, are similar to WHO grade IV GBM. Pediatric glioma primarily arises in the pons, supratentorial locations and in midline structures (thalamus, cerebellum and spinal cord)9. The specific histopathological features of WHO grade IV GBM are shown in Figure 2. The location and size of glial tumors determines the clinical presentation. Supratentorial glioblastoma multiforme (GBM) in different parts of brain is displayed in Figure 3. This analytical factor plays a paramount role in the monitoring and implementation of an appropriate treatment regimen. Commonly reported symptoms for tumors located in or subjacent to cortical regions are headache or seizures or focal neurological alterations causing hemiparesis/hemiplegia or visuospatial alteration. An increase in intracranial pressure may also be observed due to perilesional edema. Infantile spasms are also reported in 20%–30% of patients.

Histopathological examination revealing glioblastoma multiforme WHO grade IV. (A) Photomicrograph showing brisk mitotic activity (H&E, 200×). (B) Typical pallisading necrosis (H&E, 200×). (C) Glioblastoma with endothelial proliferation (H&E, 200×). (D) A case showing bizarre multinucleated tumor giant cells (H&E, 200×). (E) A case showing very high proliferation activity (Immunoreactivity to MIB-1) (IHC 200×). (F) Immunohistochemistry for p53 showing strong nuclear immunoreactivity (IHC 200×).

(A) Locations of supratentorial GBM-frontal lobe (a–c), temporal lobe (d), parietal lobe (e) and parieto-occipital region (f). (B)Locations of supratentorial GBM-temporo-parietal region (a), perisylvian (b, c), thalamus (d) and corpus callosum (e).
Anaplastic astrocytoma is characterized by tentacle-like projections towards surrounding tissue, which inhibits complete surgical excision. GBM comprises cysts, calcium deposits and blood vessels. In tuberous sclerosis complex (TSC) patients, static tubers are commonly found in the cortical parenchyma. Cortical tubers are nearly always benign hamartomas but are thought to elevate the rate of epilepsy in TSC patients. Based on MRI imaging, most TSC patients have subependymal nodules lining the ventricles. A successive neuroimaging technique helps to demonstrate succession from subependymal nodules to astrocytoma (SEGA). SEGAs exhibit both glial and neuronal features. Ultrastructure and immunophenotype studies have provided evidence of both neuronal and astrocytic differentiation. Glial fibrillary astrocytic protein (GFAP) expression is diffuse or focal compared to S100 protein expression. Neuronal markers (neuron-specific enolase (NSE) or neuron-associated cytoskeletal proteins such as β tubulin) and synaptophysin demonstrate focal positivity for some cells. However, many cells fail to be stained for either neuronal or glial markers17,18.
Molecular transformations as driving forces in glial tumorigenesis
The risk factors influencing the genesis of glioma include exposure to toxins such as vinyl chloride, ionizing radiation, electromagnetic radiation, infection with simian virus 40, gene linked ailments (Li-Fraumeni syndrome, Turcot's syndrome, and tuberous sclerosis) and chromosomal changes (chromosome 17, 7, 4 and 9). The literature also indicates that several genes such as TP53, PTEN, CDKN2A, and EGFR, etc are primarily mutated in glial tumorigenesis. It has been observed that mutation of TP53 occurs in astrocytoma, whereas amplification of EGFR and mutation of PTEN are the distinguishing features of high-grade gliomas19. Multiple genetic mechanisms generate numerous mutations, which facilitate therapeutic resistance in the tumor cells via various signaling pathways. HGGs have a high mutational burden, and their frequency indicates differential selective pressure between different locations of brain such as in the cortical region (histone H3.3 G34R or G34V mutations and BRAF-V600E mutations), midline region (histone H3 K27M mutations), pontine region (activin receptor type 1 (ACVR1) mutations) and thalamus [fibroblast growth factor receptor 1 (FGFR1) mutations]. The characteristic features of pediatric HGGs are complex genomic signatures, with significant copy number alterations (CNAs), single nucleotide variants (SNVs) and structural variants and a dorsal exophytic component mainly harboring BRAF-KIAA1549 gene fusions [in diffuse intrinsic pontine glioma (DIPG)]. The inherited predisposition factors in pediatric HGG are as follows: germline mutations in tumor suppressor genes TP53 and neurofibromin 1 (NF1); oncogenic NTRK fusions [tropomyosin 3 (TPM3)-NTRK1 and BTBD1-NTRK3]; PDGFRA mutation; EGFR mutation; focal amplification of CDK4, CDK6, cyclin D1 (CCND1), CCND2, or CCND3; histone H3.1 mutation; and K27M mutation. Thus, these mutants might be exploited as therapeutic targets in pediatric HGGs16. The major determinants of glial tumorigenesis are outlined below.
TSC manifestation in gliomagenesis
The TSC1/TSC2 complex performs a pivotal role in cortical evolution and growth regulation. A precise interface between TSC1 and TSC2 has a critical role in the development of the CNS, including morphogenesis, cell adhesion/migration and cell fate determination17. The detailed genetic arrangement of TSC1 and TSC2 is depicted in Figure 4. Mutational hotspots are absent from TSC1 or TSC2 genes.

Précis of signaling pathways involved in glioma and inhibition by mTOR, AKT, PI3K and ERK inhibitors and genetic configuration of TSC1 and TSC2. Continuous lines with arrow end exhibits activation and with blunt end exhibits inhibition. Growth factors up on binding to transmembrane receptors result in PI3Kinase activity which elevates PIP3 levels, thus activating AKT leading to anti-apoptotic/pro-cell proliferation effects. It has been also reported that HSP90 also phosphorylates AKT. AKT and /or ERK upon activation inhibits TSC1/TSC2 complex. But PTEN negatively regulates AKT. The C terminal GAP of tuberin inhibits Rheb (G protein, an activator of mTORC1) leading to increase in levels of ribosomal S6-kinase and phosphorylated ribosomal S6. Drugs potently inhibiting at different level of the signaling pathway has been also presented, respectively. The TSC1 gene comprises of 23 exons, 1164 amino acids (aa) with 130 kDa molecular mass and interacts with TSC2 in the region of 302–430 aa. It has coiled-coil (CC, aa 719–998) and potential transmembrane (TM, aa 127–144) domains at the N- and C-terminal regions. The TSC2 gene comprises of 41 exons, 1807 aa with 200 kDa molecular mass and interacts with TSC1 in the region of 1-418 aa. The gene also consists of two coiled-coils (CC, aa 346–371 and aa 1008–1021), a leucine zipper (LZ, aa 75–107), a Rheb-GAP (aa 1517–1674) and a calmodulin-binding (CAM, aa 1740–1758) domain. The N-terminal CC domain is essential for its association with TSC1. At specific aa residues the activity of TSC1 and TSC2 is synchronized by both inhibitory and activating phosphorylation events. In TSC1, the presence of glycogen synthase 3 beta (GSK3B) sites Thr357 and Thr390 activates and presence of cyclin dependent kinase (CDK1) sites Thr417, Ser584, and Thr1047 inhibits TSC1-TSC2 complex activity. In TSC2, the presence of AMP kinase (AMPK) sites Thr1227 and Ser1345 activates and presence of extracellular-related kinase (ERK2) sites Ser664; AKT/protein kinase B (PKB) sites Ser939, Ser981, Thr1462; mitogen-activated protein kinase- activated protein kinase 2 (MK2) site Ser1210 and p90 ribosomal S6 kinase 1 (RSK1) site Ser1798 inhibits TSC1-TSC2 complex activity.
TSC has a high penetrance induced by mutations and variability in TSC1/TSC2 tumor suppressor genes, which is responsible for more than 50% of deaths among children diagnosed with a brain tumor17. Distinctive TSC brain lesions comprise cortical tubers, SENs, and SEGAs20. In more than 90% of patients, SENs appear as tiny asymptomatic, intraventricular calcified protrusions in the lateral ventricles or proximate to the caudate nucleus. SENs positioned in the region of the Monro foramina can grow and transform into a SEGA. It has also been reported that solitary SEGAs even appear in the absence of any other TSC-related lesions7,18. Multiple signaling cascades are involved in the focal abnormalities of different organs due to depletion in either of the TSC1/TSC2 gene's second allele. These cascades culminate to control serine/threonine kinase mTOR, a critical regulator of many important cellular processes as depicted in Figure 4. Thus, mTORC1 inhibitors may potentially have a novel therapeutic role in the treatment of TSC patients20.
Aspect of histone mutation in glioma
The interplay between genetic and epigenetic events implies that there is a mechanism behind the epigenetic alterations (histone mutation) in glioma. One study laid the groundwork for a focus on chromatin remodeling machinery. The existence of alternative lengthening of telomeres and explicit gene expression profiles associated with H3F3A/ATRX-DAXX (α-thalassemia/mental retardation syndrome X-linked-death-domain associated protein)/TP53 mutations have also been reported. The literature reports somatic mutations in 44% of tumor cases (site of mutation ∼H3.3-ATRX-DAXX chromatin remodeling pathway) and recurrent mutations in 31% of tumors (site of mutation ∼H3F3A) with amino acid substitutions at K27 or G34 and in H3.1 histone genes HIST1H3B and HIST1H3C4. A sequencing study revealed that this mutation targets key sites on the histone tail for post-translational modifications. Hence, pharmacologic inhibition of histone demethylation might help in glioma management21,22.
Significance of IDH mutation in glioma
The involvement of isocitrate dehydrogenase (IDH) genes in the molecular pathogenesis of glioma and their translational relevance with respect to IDH mutations is considered to be a putative prognostic marker in WHO grade III gliomas and GBMs. A genome wide analysis predicted that nearly 12% of glioma patients display somatic mutations at codon 132 of the IDH gene. IDH1 or IDH2 is usually mutated in WHO grade II/III glioma or secondary GBM patients. Mutation of NADP+-dependent IDH encoded by IDH1 and IDH2 occurs in patients who develop secondary GBM from low-grade glioma19. The important discovery of IDH mutations in glioma using next-generation sequencing for glioma as well as other human diseases has elucidated the diagnostic and prognostic significance of IDH mutations in neuro-oncology.
The candidate genes IDH1 on chromosome 2q33.3 (codon R172) and IDH2 on 15q26.1 associated with GBM are mutually exclusive. Histidine change has been observed in more than 90% of IDH1 mutations at R132 codon. IDH1-associated gliomas are located in the frontal lobe (73.5% of cases) and temporal lobe (41.78% of cases). Thus, the assessment of IDH mutation is of great diagnostic relevance (immunohistochemical evaluation of anti-mIDH1R132H), prognostic significance (longer survival of GBM, grade III astrocytoma and oligodendroglioma patients) and therapeutic impact (IDH mutants possess enhanced therapeutic sensitivity; D-2-HG, an oncometabolite of mutant IDH enzymes, is a candidate for glioma therapy). Designing inhibitors of IDH mutant proteins that also penetrate the BBB might also aid in glioma stratification19,23.
FGFR-TACC fusions: a novel mutation
Chromosomal rearrangements (translocations) result into gene fusion by fusing two separate genes to produce a new gene with oncogenic properties. Accumulating evidence has shown that drugs specifically targeting oncogenic fusion proteins have therapeutic success in leukemia and lung cancer. A particular study that sought to identify oncogenic gene fusions associated with GBM development demonstrated FGFR-TACC (fibroblast growth factor receptor-transforming acidic coiled coil) fusions, the first example of a dominant mutation responsible for aneuploidy in human cancer. It has also been found that, during mitosis, FGFR-TACC fusions trigger aberrant chromosome segregation, initiating chromosome instability (CIN) and aneuploidy, the two hallmarks of cancer. FGFR-TACC fusions have been frequently identified in pediatric and adult glioma, bladder carcinoma, squamous lung carcinoma and head and neck carcinoma24,25,26,27,28,29. Since GBM is a markedly heterogeneous tumor, it is essential to determine whether such heterogeneity is also present in gliomas harboring FGFR-TACC translocations. This is reminiscent of other chromosomal translocations (BCR-ABL, EML4-ALK) and compatible with FGFR-TACC fusions in glioma24. This behavior is essential for tumor maintenance, irrespective of secondary genetic alterations that occur during tumor progression. The structural heterogeneity of FGFR3-TACC3 fusions is more distinct at the genomic level, and each fusion event signifies genomic breakpoints for identical fusion transcripts. FGFR3-TACC3 positive samples harbor small, intragenic micro-amplification events classically incorporating only the exons of FGFR3 and TACC3 genes involved in the breakpoint24,30.
A screening of glioma datasets confirmed that FGFR-TACC rearrangements occur in ∼3% of GBM and revealed the presence of FGFR-TACC fusions in IDH wild-type lower grade glioma (grade II-III) subgroups31. The results even demonstrated that in addition to mutual exclusivity between IDH1 mutations and FGFR-TACC fusions, patients with FGFR3-TACC3 rearrangements lack EGFR amplification and EGFRvIII. FGFR-TACC fusions involve the tyrosine kinase (TK) domain of FGFR and the coiled-coil domain of TACC proteins. A study involving tumor dependency on FGFR-TACC fusions in preclinical mouse models highlighted the anti-tumor effects of FGFR inhibition (AZD4547 and JNJ-42756493)32,33. The targeted inhibition of FGFR-TK in preselected IDH wild-type FGFR-TACC-positive glioma may provide clinical benefits for recurrent glioma patients. These findings offer glioma scientists a better understanding of chromosomal instability in tumors and a novel therapeutic target.
Role of infections in gliomagenesis
The role of infections in gliomagenesis has always been questionable, and viral infections have been suspected of potentially being associated with glioma risk. These uncertainties necessitate the epidemiologic investigation of the role of viral infections in glioma etiology. Cytomegalovirus (CMV), a type of herpes virus, has been found in cancerous tumors. Recently, it has been postulated that CMV infection and GBM incidence are inversely associated with socioeconomic status34,35. The association between CMV and GBM needs to be unraveled. Pundole et al critically reviewed the association between varicella zoster virus (VZV) immunity and glioma risk. Their study emphasized the comparison of VZV infection and immunity biomarkers with anti-VZV IgG levels for further studies. This neurotropic virus usually invades the host's dorsal root ganglia and induces alterations in the seroprevalence of VZV proteins (VZV ORF2p and IE63 proteins)36. Further comprehensive investigation of viral DNA, protein and RNA transcripts and cell-mediated immunity markers is essential to untangle the association between infections and glial tumorigenesis.
Challenges in glioma management: heterogeneity and recurrence in tumor microenvironment
GBM is one of the most malignant and invasive types of brain tumor. The cells actively migrate from the primary tumor site to narrow spaces within the brain. Indeed, before diagnosis, the single tumor cells may potentially create a hub in the brain. Usually, cancer patients have tumor tissue and normal tissue, but in the brain, there is a blend of normal and cancer cells. This is the most basic and confounding parameter for glioma therapeutics. Thus, conventional therapeutic strategies are not successful in the treatment of GBM, resulting in poor survival rates. The underlying reason for this ineffectiveness might be because of our superficial scientific and clinical approaches. The mainstream treatment for glioma revolves around surgery, radiation and chemotherapy. Tumor locations near eloquent sites and the infiltrative nature of glioma reduce the likelihood of complete surgical abscission of a tumor mass. Radiation therapy is employed in combination with surgical resection but is limited since the tumor center is hypoxic, and the presence of oxygen is essential for effective radiation therapy. Furthermore, tumor recurrence and radioresistance limit the effectiveness of radiation therapeutic approaches. The insensitivity of glioma cells to chemotherapeutic agents, the inability of such agents to breach the BBB, and the expulsion of such agents from cells due to multidrug-resistant protein expression restrain the prevailing chemotherapeutic strategies. Moreover, radiation and chemotherapy lead to short-term memory deficit, physical fatigue, and weakness37.
The clinical trial hurdles that impede the development of glioma therapies include the following:
usually patients are not enrolled (fewer glioma patients than those with other tumors);
the period between glioma diagnosis and clinical response dominates the disease prognosis rate;
patients undergoing surgical debulking and external beam radiation are preferred.
Conventional therapy results in median survival of only 10 to 12 months, and hence, it is rational to start with an investigational approach.
Modeling glioma in animals would aid in the identification of the genetic proceedings and molecular mechanisms contributing to tumorigenesis within the CNS and in the evaluation of potential therapeutic strategies38. The factors responsible for the failure of in vivo studies of glioma include the following:
The glioma models fail to reflect the biological properties of humans;
The pharmacokinetic profiles vary between the animals used and humans;
The tumors established differ from humans in terms of cellular heterogeneity.
Tumor heterogeneity
GBM comprises pathological and phenotypic blends of cells exhibiting cellular and nuclear polymorphism. The heterogeneous nature manifests as mixed cytological subtypes, regional differences in gene expression, and non-uniform representations of key gene mutations and genomic alterations39,40,41. Whether the inherent interactivity between tumor cells, genomic instability, or stochastic noise at the level of transcription, translation, or post-translational modifications has any influence on intratumoral heterogeneity has yet to be unraveled. An examination of dynamic heterogeneity at the cellular level is essential for understanding the origin of cells, potential therapeutic targets and source of tumor recurrence as well as for the identification of optimal cell-specific therapies. Recently, single-cell RNA-sequencing methods have confirmed intratumoral heterogeneity with different morphological, self-renewal and proliferative capacities. Differing treatment responses based on patient-specific dynamics have also been reported. Clonal evolution, CSCs and interclonal cooperativity promote tumor evolution and heterogeneity. Heterogeneity contributes to the failure of targeted therapy owing to the survival of genetically mutated heterogeneous populations of malignant cells. Tumoral heterogenic patterns might stratify patients individually, enabling the selection of appropriate therapeutics. Hence, intra-tumoral heterogeneity significantly contributes to the development of prognostic/predictive biomarkers and personalized treatment regimens42,43,44,45,46.
Tumor recurrence
The high propensity for tumor recurrence is the critical parameter responsible for unfavorable prognosis in glioma. The challenges of recurrent GBM are: 1) uniform definition and criteria regarding recurrence is indefinite due to newly formed lesions and infiltrative nature; 2) institutional variation in therapeutic strategy and 3) tumor heterogeneity. Recurrence often occurs as a local continuous growth within 2–3 cm of the lesion margin, at the original tumor site, through newly formed parenchymal lesions, or as unusual relapse patterns in midline tumors47. GBM recurrence after treatment occurs either from the bulk of the mass or within 20 mm of its boundary as detected by T1-weighted MR imaging (∼97% of cases)48. Gadolinium-enhanced MR imaging, PET and MR spectroscopy are also used in surveillance of recurrent GBM47. PET demonstrates that high regional glucose metabolism correlates with cellularity, patient survival and radiation necrosis49. MR spectroscopy discriminates between localized radiation necrosis and recurrent tumors through high Cho levels50.
Current therapeutic strategies and their limitations in glioma treatment
Glioma therapy involves multidisciplinary approaches comprising treatment, diagnosis and monitoring of aggressive malignant states. In low-grade tumors, the possibility of recurrence should be monitored, and in high-grade tumors, differential recurrence resulting from treatment-instigated alterations (radiation necrosis) should be monitored. For TSC individuals <20 years, age-dependent monitoring should be performed every 2 years. Stable glial tumors require no monitoring, but growing glial tumors require continuous monitoring. Tumors >1 cm require MRI scanning every 6 months. During pre- and post-treatment, neuroimaging techniques are used to diagnose and examine the site, extent and biological activity of the tumor51. Different neuroimaging techniques that are used for glioma are listed in Table 3.
The early detection of tumors is subtle due to a lack of precise symptoms. Patients usually report positional headache (worse in a dependent position), visual obscurations, exacerbation of focal symptoms, or sudden aggravation of seizures possibly followed by lethargy, nausea, vomiting, and diplopia18,52. Conventional treatment regimens revolve around surgery, radiation therapy and chemotherapy. These treatment strategies are inadequate in comparison with the versatile avenues of cancer progression. Typically, intermittent neuroimaging and surgical abscission enable glioma management. Hydrocephalus may be easily avoided by early surgical intervention. Surgical resection is usually unsuccessful due to regrowth of the tumor, acute morbidity and the need for ventriculoperitoneal shunting. Surgical excision is generally followed by fractionated radiotherapy (up to 54 Gy)54. Radiation therapy (RT) in combination with surgery has shown better results for glioma control, but the drawbacks of RT include damage to adjacent normal tissues and acquired radioresistance. Selecting an appropriate medical regimen for glioma is difficult, particularly when the question arises amid surgery and chemotherapy. Gliomas exhibit high VEGF and dense vasculature. Most of the astrocytic tumor cells show an elevated level of indicators of mTOR activation (phospho-S6K, phospho-S6, and phospho-Stat3)18, which are also the cause of tumor proliferation and energy metabolism. The first chemotherapy substitute for surgery for tumors was launched in a recent clinical study of the function of angiogenic and mTOR inhibitors in inducing regression of glioma and astrocytoma associated with TSC55. Regardless of continuous advancements in chemotherapy, bypassing the BBB and acquired resistance due to transporter protein up-regulation in cancer stem cells are the key hurdles56.
Surgery
For nearly all glioma patients, surgery is considered the benchmark for restoring and relieving the symptoms of mass effect. Neuroimaging confirmation of tumor progression and symptoms of increased intracranial pressure are the indications for surgical resection. The different indications for surgery in glioma are listed in Table 4. In a case of cerebellopontine angle (CPA) tumor with right-sided ventriculoperitoneal shunt, a subtotal tumor was excised by employing a left retromastoid suboccipital approach. The respective images are depicted in Figure 5. A neurosurgeon's perspective regarding surgical removal of the tumor relies upon the following four parameters: the nature of the lesion; neurological condition of the patient/Karnofsky performance status; arresting tumor growth; and arresting malignant transformation7,61. Various technical aids, such as neuronavigation and intraoperative MRI (iMRI), can be used to maximize the extent of resection in gliomas. Resective surgery for malignant glioma aids in decompressing tumor bulk, relieving pressure (vital for neurological improvement), reducing neoplasm volume (enhances adjuvant postoperative management), and defining a specific histopathological diagnosis (for selecting an appropriate therapy and predictive prognosis)47.

A surgical case displaying left CPA tumor (A) – right sided ventriculoperitoneal shunt (B) followed by subtotal excision of tumor via left retromastoid suboccipital approach (C).
Radiation therapy
Radiation therapy (RT) is usually implemented after surgery to treat tumors in vulnerable sites and for recurrent gliomas. A large randomized trial showed an increase in time to progression after early RT compared to RT at the time of progression62. Early RT (dose of 54 Gy in fractions of 1.8 Gy) improved median progression free survival from 3.4 to 5.3 years, indicating that the timing of RT is less relevant as long as it is given63. Reirradiation is frequently employed in recurrent glioma64. Fractionated stereotactic radiotherapy also benefits recurrent GBM patients65,66. Radiation oncologists hesitate to re-treat local recurrences of GBM because of the inability to regenerate or restore CNS tissues after radiation injury. A significant restoration of critical CNS structures has been observed with the use of modern high-precision radiotherapy equipment and enhanced imaging techniques. To limit the exposure of normal brain tissue outside the intended treatment area and to deliver very high doses of focused radiation, intensity modulated radiation therapy (IMRT), stereotactic RT, gamma knife, cyber knife and proton beam techniques are being employed60. However, RT is palliative because of radioresistance. The clinical response assessment criteria for glioma (disease progression and response) on the basis of the MacDonald and Response Evaluation in Solid Tumors (RECIST) 1.1 criteria comparison has validated the one-dimensional approach for solid tumor measurement and addressed the key issues for partly necrotic tumors and distinct cystic lesions67.
The following have been proposed as possible mechanisms underlying radioresistance in glioma68,69,70,71,72,73:
-
Increased DNA damage response
-
Differential cyclooxygenase response
-
Elevated HSP 70 and 90
-
Increase in DNA double strand breakage reassembly in association with micronuclei
-
Varying interferon-β response
-
Divergent cell cycle arresting patterns
-
Modulating cyclin-dependent kinase inhibitor expression and autophagy
-
BCL-family protein modulation
-
Aberrant p21 regulation in wild-type p53 radioresistant GBM cells
-
Enriched CD133 (Prominin-1) marker
-
Failure of p53 to induce p21bax expression
-
Wnt activation
-
Alteration in Notch signaling
-
Radiosensitivity critically regulated by various kinases (Akt, BCR-ABL, EGFR, Erb-B2, VEGFR2)
-
Ionizing radiation enhancing MMP-2 secretion, leading to increased invasiveness and malignancy of glioma cells.
Chemotherapy
Chemotherapy is an important adjuvant to radiotherapy following surgical resection of gliomas. The growth factors that play a pivotal role in brain tumor development are platelet-derived growth factor (PDGF), epidermal growth factor (EGF), insulin-like growth factors (IGFs), fibroblast growth factor 2 (FGF2), ciliary neurotrophic factor (CNTF), hepatocyte growth factor/scatter factor (HGF/SF), vascular endothelial growth factor (VEGF) and transforming growth factor-β (TGF-β). EGF and other ligands such as TGF-α activate the members of the EGF receptor family (ErbB/HER1-4). In gliomas, EGFR (HER1 or c-erbB1) is the most studied receptor. Overexpression of EGFR has been found in approximately half of GBMs, and approximately 40% of GBMS have EGFR deletions. Molecular and genetic anomalies and involvement of growth factors in gliomagenesis is depicted in Figure 1. In newly diagnosed GBM cases, temozolomide (TMZ) adjuvant to RT has clinically and statistically significant effects on survival without affecting toxicity levels59,74. The different growth factors involved in gliomagenesis discussed in Table 5, the signaling pathways involved in glioma, and the roles of AKT, PI3K, ERK and mTOR in Figure 4 indicate that Akt, PI3K, ERK and mTOR inhibition might be promising targets for glioma treatment. Deletion of NFKBIA (encoding nuclear factor of K-light polypeptide gene enhancer in B-cell inhibitor-α), an EGFR inhibitor signaling cascade, promotes glial tumorigenesis but does not induce any EGFR alterations. Deletion of NFKBIA and amplification of EGFR actually show a pattern of mutual exclusivity86. Notch signaling influence on brain CSC's and the key role of these tumor-initiating cells in glioma maintenance indicates that targeting these cells by Notch cascade inhibition may be worth further investigation87. Additionally, MMP inhibition may also be a potential antiangiogenic therapeutic modality. The chemical structure, mode of action and effect on glioma of different EGFR, VEGF, PDGF, PI3K/AKT, mTOR and MMP inhibitors are listed in Table 6.
TMZ is one of the leading compounds in glioma chemotherapy. It is an alkylating agent that potentially enters the CSF, bypassing hepatic metabolism for activation with predictable bioavailability and minimal toxicity. It has been approved in the US for refractory anaplastic astrocytoma and in the EU for recurrent tumors. TMZ administration in both concomitant and adjuvant phases prolongs survival and delays progression97. Time to progression and QoL benefits have been observed in recurrent glioma cases98. In an evidence-based clinical study, Olson et al recommends TMZ over procarbazine for first relapse of GBM99. In a randomized phase III trial by Stupp et al, a TMZ+radiation regimen proved to be a statistically significant and clinically meaningful therapy, with a median follow-up of more than 5 years100.
However, the major constraints of chemotherapy are bypassing the BBB, its interaction with anti-seizure medications and/or steroids, intrinsic or acquired resistance, and cases of recurrent glioma. Bevacizumab (an anti-VEGF inhibitor) and bevacizumab+irinotecan/etoposide/CCNU are employed for recurrent glioma. Other agents that have been tested for recurrent GBM are cediranib (pan-VEGFR), erlotinib/gefitinib (EGFR), cilengitide (α and β integrins), rindopepimut (EGFRvIII), vorinostat (HDAC), XL-184 (EGFR, C-MET), Tipifarnib (farnesyltransferase), enzastaurin (PKC) and temsirolimus (mTOR)101.
Combination therapy
Human malignant gliomas seldom show any dependency on a single oncogene or tumor suppressor. This might be responsible for the failure of agents targeting only one oncogenic pathway in clinical trials. It has also been revealed that EGFR pathway hyperactivation is associated with resistance to treatment with RT and chemotherapy102. There are two important considerations that effect glioma therapy; first, numerous RTKs are co-activated in glioma cells103; and second, issues of acquired resistance. Thus, a combination of surgery, chemotherapy and RT are essential for sensitizing the glioma cells to efficient therapeutics. Combination regimens for glioma are listed in Table S1. Furthermore, an approach involving a combination of different molecular-targeted agents with standard cytotoxic agents has yet to be developed104.
Next generation therapeutic strategies to combat glioma
Despite various treatment modalities, such as surgery/RT/chemotherapy and their prevailing limitations, glioma patients have a dismal prognosis. A different approach to recurrent GBM therapy uses medium frequency electrical fields. In 2011, a novel device NovoTTF100A (Novocure, New Hampshire, USA) was employed for arresting dividing cells in mitosis. The device was as effective as chemotherapy, and quality of life was better compared to systemic therapy. Researchers also found that the device might aid in potentiating the effects of chemotherapy (www.clinicaltrials.gov, NCT00916409).
Progress in glioma therapy could be attained by improved comprehension of glioma biology, identification of relevant targets and signaling pathways for treatment interventions, development of personalized medicine, optimization of surgery and RT, and innovative neuroimaging techniques. Proteogenomic characterization is a potential strategy that could lead to identification of molecular drivers, molecular classification of disease subgroups and glioma treatment. The ultimate goal of targeted therapy should be “selectivity,” ie. inhibiting only tumor cells. The targeted approaches currently in clinical trials or in laboratory development include drugs, monoclonal antibodies, immunotherapy, small molecules inhibiting specific proteins and specific targeting of CSC's. Thus, there is a need for unconventional treatment strategies to curb glioma. Strategies such as gene therapy, microRNA (miRNA) therapy, stem cells, and immunotherapy may potentially lead to effective GBM treatments.
Proteogenomic characterization of glioma
Next-generation sequencing is being widely employed to characterize developed genomic and transcriptome alterations in human diseases. The insights provided by omics studies in search of protein signatures and biomarkers for glioma that highlight the expression of specific proteins in different grades of glioma have been already discussed in Table 2. Transcriptome profiling for gene expression fails to correlate with protein expression and posttranslational modifications (PTMs). Hence, advancements in proteomic platforms with inclusive proteome arrays would aid in providing systematic and analogous protein expression evidence that is complementary to DNA and RNA profiles110. An initiative integrating genomic, proteomic, and phosphoproteomic dimensions might aid in identifying differential signaling pathways and functional modules exhibiting substantial associations with patient outcomes. Such methods would likely identify PTMs, revealing a strong association between histone acetylation and the homologous recombination deficiency (HRD) phenotype111.
Recently, Zhang et al provided a comprehensive analysis of the molecular components and underlying mechanisms of ovarian cancer. They performed an inclusive mass-spectrometry-based proteomic characterization of 174 ovarian tumors of the high-grade serous carcinoma (HGSC) category. The insights provided by the study include the following: the influence of different copy-number alternations on the proteome; proteins associated with chromosomal instability; specific protein acetylations associated with HRD; the influence of the somatic genome on the cancer proteome; and associations between proteins and PTM levels and corresponding clinical outcomes in HGSC111. The complex proteome analysis was primarily carried out through a mass spectrometry (MS)-based shotgun proteomics approach. The resultant peptide mixtures obtained from protease-digested complex protein samples were fractionated on HPLC columns, followed by tandem MS analysis. The subsequent MS/MS spectra were compared to a protein database for protein identification and PTMs. A study of Alzheimer's disease (AD) by Wang et al highlighted that gas-phase fractionation of peptide ions enhanced peptide identification by ∼10%. The identification of 96 127 peptides and 10,544 proteins at a 1% protein false discovery rate was enabled by combining basic pH liquid chromatography (LC) prefractionation with a long gradient LC-MS/MS platform110. This study contributed to the systematic optimization of long gradient chromatography MS for a profound study of the brain proteome. Li et al used proteogenomics to improve gene annotation and interpretation of proteomics data. They employed an integrative proteogenomics pipeline JUMPg for processing a label-free MS data set of AD, recognizing 496 new peptides (amino acid substitutions; alternative splicing; frame-shift; non-coding gene translation), and analyzing a stable-isotope-labeled data set of multiple myeloma cells, revealing 991 sample-specific peptides (protein sequences in the immunoglobulin light chain variable region). The multistage strategy included a modified database structure, tag-based database probe, peptide-spectrum match sieving, and data conception. Their study highlighted expression of a novel protein PNMA6BL in the brain and the use of the JUMPg program in proteogenomics for multi-omic data integration112.
Thus, understanding the molecular basis of glioma can be enhanced by an in-depth evaluation of pathway activity by using a proteogenomic approach to show the correlation between genotype and proteotype and ultimately clinical phenotype.
Gene therapy
In brain tumors, gene therapy transfers genetic material into the tumor cells. Gene therapy has the ability to target invasive tumor cells resistant to conventional therapy. The different gene therapy strategies for glioma include the following: (a) Suicide gene therapy - DNA synthesis is terminated by a prodrug-activating gene. This method results in gene expression for a shorter period and enhanced sensitivity, but in vivo, the gene transfer rate is poor and fails to target dispersed tumor cells. (b) Oncolytic viral therapy – viral replication lyses tumor cells. For this method, transduction efficiency and viral titers are high, but there is a possibility of host immune rejection, and local administration during surgery is required. (c) Immunomodulatory therapy – involves stimulation of an antiglioma immune response and regulation of the tumor microenvironment. However, the limitations are immunosuppression and lack of antigen-presenting dendritic cells. (d) Synthetic vectors (nanoparticles) – is a safer approach for silencing gliomagenesis genes by RNAi and siRNA delivery. A sustained release pattern is an added advantage, but reduced intratumoral distribution and transduction efficiency have also been observed.
Gene therapy has demonstrated significant therapeutic efficacy in preclinical and phase I trials but has failed in phase III trials because of the heterogeneity and invasiveness of GBM, anatomical features of the CNS, host immune system and inadequacy of GBM animal models113.
miRNAs-anti-oncogenic therapy
The 'oncomirs' or miRNAs have been found to be associated with different human cancers and are also viewed as promising therapeutic targets in cancers. Some miRNAs show oncogenic activity by upregulating miRNAs in cancer and targeting tumor suppressor genes, while others illustrate tumor suppressor activity by downregulating miRNAs in cancer. This distinct activity of miRNAs depends on the biological context and tissue type. The potential role of miRNAs in CSCs, to curb resistance, has been described in recently published studies. In GBM, several miRNAs regulating oncogenic and tumor suppressor proteins have been identified. Identification of dysregulated miRNAs in GBM that are potential participants in glioma genesis, such as miR-21, miR-196, miR-10b, miR-128-1, and miR128-2, has led to more accurate predictions of clinical outcome than mRNA profiles. A stronger correlation between clinical outcome and miRNA-mRNA expression signature has also been acknowledged114,115.
Cancer Stem Cell (CSC) therapy
Research regarding CSCs and their role in GBM survival and relapse is being carried out on a larger scale. It seems that the heterogeneity of brain tumors is dependent on the heterogeneity of their CSCs, and their involvement in complex mechanisms largely depends upon their microenvironment. Consequently, CSCs could also be a potential therapeutic target in GBM. According to Binello and Germano, direct targeting refers to augmenting CSC functions via EGFR/PI3K/Akt inhibition and inducing differentiation to curb resistance to standard treatments, whereas indirect targeting addresses perivascular niches, hypoxic niches and immune evasion. The molecular characteristics exhibited by CSCs include the expression of multidrug-resistance genes (such as ABCG2 and BCRP1) and the promotion of drug efflux and CSC survival. In GBM-tumor sphere cells, the expression of multidrug resistance genes has been found to be enhanced. Expression of the stem cell-associated protein CD133 helps in the identification and isolation of GBM CSCs. It has been recently determined that CD133+ GBM cells are more radioresistant than CD133− cells75. In spite of having an intact G2 checkpoint, CD133+ cells lack the intra-S-phase checkpoint. Compared to GBM cell lines, in vitro CD133+ GBM CSCs are more radiosensitive with a reduced capacity to repair DNA double-strand breaks 116. Hedgehog-Gli signaling inhibitors have been used to treat tumors and are associated with CSCs and the regulation of proliferating CSCs117. These inhibitors induce GBM-derived neurosphere cells to lose their tumorigenicity, reduce stem cell marker expression and target radiation to unaffected GBM cells. Therefore, Hedgehog blockade potentially offers a new therapeutic in combination with chemotherapy or RT.
Akt inhibitors play a substantial role by sensitizing brain CSCs to radiation for inducing apoptosis and directly targeting CSCs. The CSCs of the brain are maintained within vascular niches that imitate neural stem cell niches118. Thus, selective annihilation of CSCs could be achieved by employing a combination of chemotherapy and RT with antiangiogenic drugs.
Immunotherapy
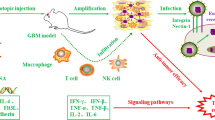
Immunotherapy provides a durable and targeted treatment against cancer by harnessing the body's adaptive immune mechanisms. The principal mechanisms employed are the improvement of the immune response and targeting of specific antigens. The immune system in the brain is highly active and interacts with brain tumors. However, the diffuse and infiltrative nature of glial tumors poses a challenge to effective immunotherapy. An invasive tumor residing behind the BBB is isolated from effective immunosurveillance and ultimately leads to “immunologically silent” tumor peninsulas. The ability of adoptively transferred T cells to migrate and mediate regression in areas of invasive GBM is unclear119.
The prevailing multimodal therapy is non-specific and is limited by tissue toxicity. In contrast, immunotherapy research has shown substantial evidence of T cells' ability to eradicate large, well-established tumors in mice and humans while sparing the normal brain cells. Glioma cells express and secrete numerous immunosuppressive molecules regulating immune cell functions. The true mechanism of immunosuppression involves a combination of factors, including regulatory T cells (Tregs), tumor-associated PD-L1 expression, and CTLA-4 signaling120,121,122. The current immunotherapeutic approaches focus on enhancing T-cell function by generally stimulating the immune system or by attacking specific tumor cell antigens. These strategies include the use of vaccines, adoptive cell transfer and immune checkpoint inhibitors.
Vaccines
The use of vaccines is an active immunotherapeutic approach that is intended to activate and expand tumor-specific T cells to induce an anti-tumor response. Various cancer vaccines are made by expanding and stimulating dendritic cells (DCs). Currently, all vaccines in clinical trials are struggling with the challenge of tolerance so that the cancer cells can be recognized by a patient's immune system. The steps involved for efficient vaccination are, first to identify the specific tumor antigens and, second, to generate an anti-tumoral response and to block all the inhibitory immune mechanisms by adopting proper immune strategies. Naive immune system vaccination is stimulated by antigen presenting cells known as dendritic cells (DCs), which help maintain self-tolerance. In vaccination studies, DCs are usually loaded with specific tumor-associated peptides, tumor RNA, cDNA, cell lysate, or apoptotic cells. DC generation, maturation, subtype, dosing, vaccination schedule, route of administration, and antigen loading approaches are the factors that must be standardized before DC vaccination can enter the clinical phase. In a phase I study of a DC vaccine in high-grade glioma, patients exhibited longer survival time, and a positive cytotoxic T-lymphocyte (CTL) response was induced. Fascinating results have been observed in patients with malignant glioma, anaplastic astrocytoma and GBM who have been vaccinated using tumor lysate as an antigen source123. An efficient depletion of tumor-specific CSCs has been observed by CTLs generated by DC vaccination+CSC derived tumor lysate in mouse glioma GL261 neurospheres (GL261-NS)124. Studies of the SOX2 gene have led to the genesis of specific CTLs against HLA-A0201-restricted SOX2-derived peptide (TLMKKDKYTL)125. This remarkable discovery helped in lysing glioma cells and developing T cell-based immunotherapy of brain CSCs.
In recurrent glioma patients, the antiEGFRvIII vaccine strategy is also being evaluated, with randomization of first or second recurrent patients to receive either bevacizumab+vaccine or placebo (bevacizumab naïve patients) or bevacizumab+vaccine (antiVEGF refractory tumors) (www.clinicaltrials.gov, NCT01498328).
Adoptive cell transfer
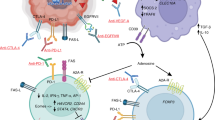
Adoptive cell transfer mainly involves the transfer of tumorigenic immune cells to cancer patients. Lymphocytes are isolated from the blood, followed by transferring molecules that recognize tumor antigens (artificial T-cell receptors) onto the lymphocytes, providing them with a new and enhanced function. This process is called the chimeric antigen receptor (CAR) method126. CAR enhances the ability of T cells to specifically target antigens and efficiently kill tumor cells by combining the specificity of an antibody and the cytotoxicity of CTL. CAR consists of single chain variable fragment (scFv) of a tumor antigen-specific antibody and the signaling domains of the T cell receptor (TCR)127. In fact, CAR bypasses the mechanisms (down-regulation of MHC and costimulatory molecules and induction of suppressive cytokine and regulatory T cells) by which tumor cells escape immunorecognition. Recently, clinical trials of CAR-mediated adoptive immunotherapy in a variety of tumor systems such as renal cell carcinoma128, indolent B-cell and mantle cell lymphoma129, neuroblastoma130, acute lymphoblastic leukemia131, and chronic lymphoid leukemia132 have established their significant potential. However, some adverse events have also been reported resulting from the administration of CAR T cells against tumor antigens that are simultaneously expressed on normal tissues133.
Accumulating evidence regarding enhanced antitumor activity due to the activation, proliferation and survival of CAR T cells comprising co-stimulatory molecules has led to new innovations in glioma therapy. The most commonly used co-stimulatory molecule is CD28134. The prerequisite for attaining a significant response in CAR-mediated immunotherapy is the CAR architecture and the choice of tumor associated antigen. EGFR variant III (EGFRvIII) is an oncogenic variant frequently expressed in glioma and other types of cancer135. EGFRvIII expression in association with survival, invasion, angiogenesis and radio/chemo resistance makes it an attractive target. Recent reports have demonstrated that systemically administered EGFRvIII+CAR T cells potentially migrate to the invasive edges of tumors, suppress tumor growth and enhance survival of intracranial D-270 MG tumor-bearing mice136.
Immune checkpoint inhibitors
Immune checkpoint activating molecules (CD200, a positive regulator of MDSC32, and the immunosuppressive Fgl2) and immune checkpoints [PDL-1 (Programmed cell death protein 1 ligand), IDO (indoleamine 2,3-dioxygenase) and CTLA-4 (Cytotoxic T-lymphocyte-associated protein 4)] are upregulated within the tumor tissue and in the serum of patients with high-grade glioma. Therefore, immune checkpoint inhibitors might play a crucial role in glioma immunotherapy. Immune checkpoint therapy has emerged as a potent addition to glioma therapy. Clinically successful checkpoint blockades such as CTLA-4 and PD-1 both alone and together have shown promising outcomes. Other targets are LAG-3, TIM-3, KIR, and GITR. Checkpoint inhibitors may be effective as monotherapy or in combination with chemotherapy and/or radiation therapy. Significant improvements in tumor regression and overall survival have been attained due to the synergy between the antibodies and either of the two conventional modalities137,138. The key immune checkpoints that play a role in gliomagenesis, such existing preclinical and clinical data, antitumor efficacy, and clinical applications for each checkpoint and in combination with chemotherapy and radiation, are listed in Table S2.
Molecular targets initiating metastasis
Glioma is among the most vascularized and invasive cancers. In GBM, angiogenesis is correlated with malignancy grading and inversely correlated with patient survival. Glioma cells infiltrate and disrupt physical barriers (such as basement membranes, extracellular matrices and cell junctions). The overexpression of several members of the zinc-based proteinase family (metalloproteinases) is a hallmark in the process of invasion. The migratory potential throughout brain structures, infiltrative nature and rapid tumor progression of glioma cells make them elusive targets for effective treatment. Moreover, inadequate results with chemotherapy have led to the study of molecules targeting specific pathways or proteins involved in glioma progression. Therefore, the migratory behavior of glioma cells could potentially be efficiently managed through the identification of the molecular targets that induce metastasis, which could be achieved through a better comprehension of glioma biology.
Concluding remarks and future directions
Tumor location, potential symptoms and the risks/benefits of the various treatment regimens (surgery/radiation/chemotherapy) are the parameters that are taken into consideration in the clinical management of glioma patients. The migratory potential of glioma cells over relatively long distances through brain structures make them elusive targets for effective surgical management. Despite the continuous development of new chemotherapeutic agents, the ability to bypass the BBB and acquired drug resistance are still constraints. Rather than attempting to control the migration of diffuse glioma, interventions that specifically target the invasive phenotype should be developed. However, recent advancements in neuroimaging will contribute to early diagnosis and initiation of glioma management. Advancements in non-invasive imaging protocols and a better understanding of glioma biology will enable neuro-oncologists to decipher the molecular, cellular, genetic and epigenetic makeup of tumors. This information might pave the way to personalized glioma therapies. Nanotechnology, transplantation of stem cells, drug dosing or timing variations, mitigation of immunosuppressive mechanisms and a better understanding of miRNA and mRNA interactions are also some of the strategies that may facilitate GBM stratification. A better understanding of the complex biology of glioma and identification of the molecular targets that initiate metastasis will facilitate the development of a novel class of anticancer therapeutics with improved efficacy and safety. Additionally, proteogenomic characterization may also identify molecular drivers and lead to molecular classification of glioma subgroups. The insights provided by omics studies will facilitate identification of glioma protein signatures and biomarkers as well as the design of potential treatment regimens. Next generation therapies comprising progressive neuroimaging techniques, termination of DNA synthesis by prodrug-activating genes, silencing gliomagenesis genes, targeting miRNA oncogenic activity, sensitizing cancer stem cells by Hedgehog-Gli/Akt inhibitors+radiation and employing tumor lysates as antigen sources for efficient depletion of tumor-specific CSCs by cytotoxic T lymphocytes along with conventional strategies will provide a new paradigm in glioma therapeutics with a focus on early diagnosis and successful management.
References
Chen L, Zou X, Wang Y, Mao Y, Zhou L . Central nervous system tumors: a single center pathology review of 34,140 cases over 60 years. BMC Clin Pathol 2013; 13: 1–14.
Ohgaki H, Kleihues P . Genetic alterations and signaling pathways in the evolution of gliomas. Cancer Sci 2009; 100: 2235–41.
Richard H, Stogner-Underwood K, Fuller C . Congenital oligodendroglioma: clinicopathologic and molecular assessment with review of the literature. Case Rep Pathol 2015; 1–4.
Chintagumpala M, Gajjar A . Brain tumors. Pediatr Clin North Am 2015; 62: 167–78.
Parish JM, Bonnin JM, Goodman JM, Cohen-Gadol AA . Intrasellar ependymoma: clinical, imaging, pathological, and surgical findings. J Clin Neurosci 2015; 22: 638–41.
Ren H, Chen XL, Sun GC, Hu S, Zheng G, Li FY, et al. Resection of subependymal giant cell astrocytoma guided by intraoperative magnetic resonance imaging and neuronavigation. Childs Nerv Syst 2013; 29: 1113–21
Beaumont TL, Limbrick DD, Smyth MD . Advances in the management of subependymal giant cell astrocytoma. Childs Nerv Syst 2012; 28: 963–8.
Adriaensen ME, Schaefer-Prokop CM, Stijnen T, Duyndam DA, Zonnenberg BA, Prokop M . Prevalence of subependymal giant cell tumors in patients with tuberous sclerosis and a review of the literature. Eur J Neurol 2009; 16: 691–6.
Hirose T, Scheithauer BW, Lopes MB, Gerber HA, Altermatt HJ, Hukee MJ, et al. Tuber and subependymal giant cell astrocytoma associated with tuberous sclerosis: an immunohistochemical, ultrastructural, and immunoelectron and microscopic study. Acta Neuropathol 1995; 90: 387–99.
Suk K . Proteomic analysis of glioma chemoresistance. Curr Neuropharmacol 2012; 10: 72–9.
Niclou SP, Fack F, Rajcevic U . Glioma proteomics: status and perspectives. J Proteomics 2010; 73: 1823–38.
Vogel TW, Zhuang Z, Li J, Okamoto H, Furuta M, Lee YS, et al. Proteins and protein pattern differences between glioma cell lines and glioblastoma multiforme. Clin Cancer Res 2005; 11: 3624–32.
Iwadate Y, Sakaida T, Hiwasa T, Nagai Y, Ishikura H, Takiguchi M, et al. Molecular classification and survival prediction in human gliomas based on proteome analysis. Cancer Res 2004; 64: 2496–501.
Zhang R, Tremblay TL, McDermid A, Thibault P, Stanimirovic D . Identification of differentially expressed proteins in human glioblastoma cell lines and tumors. Glia 2003; 42: 194–208.
Deighton RF, McGregor R, Kemp J, McCulloch J, Whittle IR . Glioma pathophysiology: insights emerging from proteomics. Brain Pathol 2010; 20: 691–703.
Jones C, Baker SJ . Unique genetic and epigenetic mechanisms driving paediatric diffuse high-grade glioma. Nat Rev Cancer 2014; 14: 651–61.
Berhouma M . Management of subependymal giant cell tumors in tuberous sclerosis complex: the neurosurgeon's perspective. World J Pediatr 2010; 6: 103–10.
Campen CJ, Porter BE . Subependymal Giant Cell Astrocytoma (SEGA) treatment update. Curr Treat Options Neurol 2011; 13: 380–5.
Ducray F, Marie Y, Sanson M . IDH1 and IDH2 mutations in gliomas. N Engl J Med 2009; 360: 1–9.
Ess KC . Tuberous sclerosis complex: everything old is new again. J Neurodev Disord 2009; 1: 141–9.
Hochart A, Escande F, Rocourt N, Grill J, Koubi-Pick V, Beaujot J, et al. Long survival in a child with a mutated K27M-H3.3 pilocytic astrocytoma. Ann Clin Transl Neurol 2015; 2: 439–43.
Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012; 482: 226–31.
Marta Mellai VC . Laura Annovazzi, Davide Schiffer . The distribution and significance of IDH mutations in gliomas. Evolution of the molecular biology of brain tumors and the therapeutic implications, INTECH; 299–342.
Singh D, Chan JM, Zoppoli P, Niola F, Sullivan R, Castano A, et al. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science 2012; 337: 1231–5.
Majewski IJ, Mittempergher L, Davidson NM, Bosma A, Willems SM, Horlings HM, et al. Identification of recurrent FGFR3 fusion genes in lung cancer through kinome-centred RNA sequencing. J Pathol 2013; 230: 270–6.
Parker BC, Annala MJ, Cogdell DE, Granberg KJ, Sun Y, Ji P, et al. The tumorigenic FGFR3-TACC3 gene fusion escapes miR-99a regulation in glioblastoma. J Clin Invest 2013; 123: 855–65.
Wu YM, Su F, Kalyana-Sundaram S, Khazanov N, Ateeq B, Cao X, et al. Identification of targetable FGFR gene fusions in diverse cancers. Cancer Discov 2013; 3: 636–47.
Zhang J, Wu G, Miller CP, Tatevossian RG, Dalton JD, Tang B, et al. Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat Genet 2013; 45: 602–12.
Capelletti M, Dodge ME, Ercan D, Hammerman PS, Park SI, Kim J, et al. Identification of recurrent FGFR3-TACC3 fusion oncogenes from lung adenocarcinoma. Clin Cancer Res 2014; 20: 6551–8.
Wang XS, Prensner JR, Chen G, Cao Q, Han B, Dhanasekaran SM, et al. An integrative approach to reveal driver gene fusions from paired-end sequencing data in cancer. Nat Biotechnol 2009; 27: 1005–11.
Sanson M, Marie Y, Paris S, Idbaih A, Laffaire J, Ducray F, et al. Isocitrate dehydrogenase 1 codon 132 mutation is an important prognostic biomarker in gliomas. J Clin Oncol 2009; 27: 4150–4.
Tabernero J, Bahleda R, Dienstmann R, Infante JR, Mita A, Italiano A, et al. Phase I dose-escalation study of JNJ-42756493, an oral pan-fibroblast growth factor receptor inhibitor, in patients with advanced solid tumors. J Clin Oncol 2015; 33: 3401–8.
Di Stefano AL, Fucci A, Frattini V, Labussiere M, Mokhtari K, Zoppoli P, et al. Detection, characterization, and inhibition of FGFR-TACC fusions in IDH wild-type glioma. Clin Cancer Res 2015; 21: 3307–17.
Cobbs CS, Harkins L, Samanta M, Gillespie GY, Bharara S, King PH, et al. Human cytomegalovirus infection and expression in human malignant glioma. Cancer Res 2002; 62: 3347–50.
Lehrer S . Cytomegalovirus infection in early childhood may be protective against glioblastoma multiforme, while later infection is a risk factor. Med Hypotheses 2012; 78: 657–8.
Pundole X, Amirian ES, Scheurer ME . Role of varicella zoster virus in glioma risk: current knowledge and future directions. OA Epidemiol 2014; 2: 1–6.
Lindsey H . Glioblastoma: zeroing in on treatment challenges. Oncol Times 2003; 25: 8–9, 12.
Huszthy PC, Daphu I, Niclou SP, Stieber D, Nigro JM . Sakariassen PØ, et al. In vivo models of primary brain tumors: pitfalls and perspectives. Neuro Oncol 2012; 135: 1–15.
Jung V, Romeike BF, Henn W, Feiden W, Moringlane JR, Zang KD, et al. Evidence of focal genetic microheterogeneity in glioblastoma multiforme by area-specific CGH on microdissected tumor cells. J Neuropathol Exp Neurol 1999; 58: 993–9.
Maher EA, Furnari FB, Bachoo RM, Rowitch DH, Louis DN, Cavenee WK, et al. Malignant glioma: genetics and biology of a grave matter. Genes Dev 2001; 15: 1311–33.
Furnari FB, Fenton T, Bachoo RM, Mukasa A, Stommel JM, Stegh A, et al. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev 2007; 21: 2683–710.
Inda MM, Bonavia R, Mukasa A, Narita Y, Sah DW, Vandenberg S, et al. Tumor heterogeneity is an active process maintained by a mutant EGFR-induced cytokine circuit in glioblastoma. Genes Dev 2010; 24: 1731–45.
Alves TR, Lima FR, Kahn SA, Lobo D, Dubois LG, Soletti R, et al. Glioblastoma cells: a heterogeneous and fatal tumor interacting with the parenchyma. Life Sci 2011; 89: 532–9.
Soeda A, Hara A, Kunisada T, Yoshimura S, Iwama T, Park DM . The evidence of glioblastoma heterogeneity. Sci Rep 2015; 5: 7979.
Parker NR, Khong P, Parkinson JF, Howell VM, Wheeler HR, et al. Molecular heterogeneity in glioblastoma: potential clinical implications. Front Oncol 2015; 5: 55.
Sottoriva A, Spiteri I, Piccirillo SG, Touloumis A, Collins VP, Marioni JC, et al. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc Natl Acad Sci U S A 2013; 110: 4009–14.
Hou LC, Veeravagu A, Hsu AR, Tse VC . Recurrent glioblastoma multiforme: a review of natural history and management options. Neurosurg Focus 2006; 20: E5.
Konishi Y, Muragaki Y, Iseki H, Mitsuhashi N, Okada Y . Patterns of intracranial glioblastoma recurrence after aggressive surgical resection and adjuvant management: retrospective analysis of 43 cases. Neurol Med Chir (Tokyo) 2012; 52: 577–86.
Ishikawa M, Kikuchi H, Miyatake S, Oda Y, Yonekura Y, Nishizawa S . Glucose consumption in recurrent gliomas. Neurosurgery 1993; 33: 28–33.
Wald LL, Nelson SJ, Day MR, Noworolski SE, Henry RG, Huhn SL, et al. Serial proton magnetic resonance spectroscopy imaging of glioblastoma multiforme after brachytherapy. J Neurosurg 1997; 87: 525–34.
Alafaci C, Granata F, Cutugno M, Caffo M, Caruso G . Modern neuroimaging techniques in the diagnosis of brain tumours. Clinical management and evolving novel therapeutic strategies for patients with brain tumors. Intech Open 2013; 55–76.
Ahmed R, Oborski MJ, Hwang M, Lieberman FS . Mountz JM, et al. Malignant gliomas: current perspectives in diagnosis, treatment, and early response assessment using advanced quantitative imaging methods. Cancer Manag Res 2014; 6: 149–70.
Nishio S, Morioka T, Suzuki S, Kira R, Mihara F, Fukui M . Subependymal giant cell astrocytoma: clinical and neuroimaging features of four cases. J Clin Neurosci 2001; 8: 31–4.
Roa W, Brasher PM, Bauman G, Anthes M, Bruera E, Chan A, et al. Abbreviated course of radiation therapy in older patients with glioblastoma multiforme: a prospective randomized clinical trial. J Clin Oncol 2004; 22: 1583–8.
Jóźwiak S, Nabbout R . Curatolo P ; participants of the TSC Consensus Meeting for SEGA and Epilepsy Management. Management of subependymal giant cell astrocytoma (SEGA) associated with tuberous sclerosis complex (TSC): clinical recommendations. Eur J Paediatr Neurol 2013; 17: 348–52.
Hide T, Takezaki T, Nakamura H, Kuratsu J, Kondo T . Brain tumor stem cells as research and treatment targets. Brain Tumor Pathol 2008; 25: 67–72.
Jiang T, Jia G, Ma Z, Luo S, Zhang Y . The diagnosis and treatment of subependymal giant cell astrocytoma combined with tuberous sclerosis. Childs Nerv Syst 2011; 27: 55–62.
Ouyang T, Zhang N, Benjamin T, Jiao J, Zhao Y, et al. participants of the TSC Consensus Meeting for SEGA and Epilepsy Management. Subependymal giant cell astrocytoma: current concepts, management, and future directions. Childs Nerv Syst 2014: 30: 561–70.
Kjellman C, Olofsson SP, Hansson O, Von Schantz T, Lindvall M, Nilsson I, et al. Expression of TGF-beta isoforms, TGF-beta receptors, and SMAD molecules at different stages of human glioma. Int J Cancer 2000; 89: 251–8.
Niyazi M, Siefert A, Schwarz SB, Ganswindt U, Kreth FW, Tonn JC, et al. Therapeutic options for recurrent malignant glioma. Radiother Oncol 2011; 98: 1–14.
de Ribaupierre S, Dorfmüller G, Bulteau C, Fohlen M, Pinard JM, Chiron C, et al. Subependymal giant-cell astrocytomas in pediatric tuberous sclerosis disease: when should we operate? Neurosurgery 2007; 60: 83–9, discussion 89–90.
van den Bent MJ, Afra D, de Witte O, Ben Hassel M, Schraub S, Hoang-Xuan K, et al. Long-term efficacy of early versus delayed radiotherapy for low-grade astrocytoma and oligodendroglioma in adults: the EORTC 22845 randomised trial. Lancet 2005; 366: 985–90.
van den Bent MJ, Snijders TJ, Bromberg JE . Current treatment of low grade gliomas. Memo 2012; 5: 223–7.
Butowski NA, Sneed PK, Chang SM . Diagnosis and treatment of recurrent high-grade astrocytoma. J Clin Oncol 2006; 24: 1273–80.
Torok JA, Wegner RE, Mintz AH, Heron DE, Burton SA . Re-irradiation with radiosurgery for recurrent glioblastoma multiforme. Technol Cancer Res Treat 2011; 10: 253–8.
Vordermark D, Kölbl O, Ruprecht K, Vince GH, Bratengeier K, Flentje M . Hypofractionated stereotactic re-irradiation: treatment option in recurrent malignant glioma. BMC Cancer 20051; 5: 55.
Upadhyay N, Waldman AD . Conventional MRI evaluation of gliomas. Br J Radiol 2011; 84: S107–11.
Wang J, Wakeman TP, Lathia JD, Hjelmeland AB, Wang XF, White RR, et al. Notch promotes radioresistance of glioma stem cells. Stem Cells 2010; 28: 17–28.
Williams, JR, Gridley, DS, Slater, JM . Radiobiology of radioresistant glioblastoma, advances in the biology, imaging and therapies for glioblastoma. Intech 2011; 22.
Shu HK, Kim MM, Chen P, Furman F, Julin CM, Israel MA . The intrinsic radioresistance of glioblastoma-derived cell lines is associated with a failure of p53 to induce p21(BAX) expression. Proc Natl Acad Sci U S A 1998; 95: 14453–8.
Kim Y, Kim KH, Lee J, Lee YA, Kim M, Lee SJ, et al. Wnt activation is implicated in glioblastoma radioresistance. Lab Invest 2012; 92: 466–73.
Milanović D, Firat E, Grosu AL, Niedermann G . Increased radio-sensitivity and radiothermosensitivity of human pancreatic MIA PaCa-2 and U251 glioblastoma cell lines treated with the novel Hsp90 inhibitor NVP-HSP990. Radiat Oncol 2013; 8: 42: 9.
Martinou M, Giannopoulou E, Malatara G, Argyriou AA, Kalofonos HP, Kardamakis D, et al. Ionizing radiation affects epidermal growth factor receptor signalling and metalloproteinase secretion in glioma cells. Cancer Genomics Proteomics 2011; 8: 33–8.
Swartling FJ . Identifying candidate genes involved in brain tumor formation. Review based on the doctoral thesis “Screening for candidate brain tumor genes – Identifying genes that cooperate with Platelet-derived growth factor in glioma development and progression. Upsala J Med Sci 2008; 113: 1–38.
Tanase CP, Enciu AM, Mihai S, Neagu AI, Calenic B, Cruceru ML . Anti-cancer therapies in high grade gliomas. Curr Proteomics 2013; 10: 246–60.
Kim RY, Mahta A, Kesari S . Glioma stem cells. InTechOpen. 271–90.
Calzolari F, Malatesta P . Recent insights into PDGF-induced gliomagenesis. Brain Pathol 2010; 20: 527–38.
Lokker NA, Sullivan CM, Hollenbach SJ, Israel MA, Giese NA . Platelet-derived growth factor (PDGF) autocrine signaling regulates survival and mitogenic pathways in glioblastoma cells: evidence that the novel PDGF-C and PDGF-D ligands may play a role in the development of brain tumors. Cancer Res 2002; 62: 3729–35.
Eyler CE, Foo WC, LaFiura KM, McLendon RE, Hjelmeland AB, Rich JN . Brain cancer stem cells display preferential sensitivity to Akt inhibition. Stem Cells 2008; 26: 3027–36.
Pal I, Mandal M . PI3K and Akt as molecular targets for cancer therapy: current clinical outcomes. Acta Pharmacol Sin 2012; 33: 1441–58.
Wander SA, Hennessy BT, Slingerland JM . Next-generation mTOR inhibitors in clinical oncology: how pathway complexity informs therapeutic strategy. J Clin Invest 2011; 121: 1231–41.
Dasgupta T, Haas-Kogan DA . The combination of novel targeted molecular agents and radiation in the treatment of pediatric gliomas. Front Oncol 2013; 3: 110.
Moavero R, Coniglio A, Garaci F, Curatolo P . Is mTOR inhibition a systemic treatment for tuberous sclerosis? Ital J Pediatr 2013; 39: 57.
Curatolo P, Moavero R . mTOR inhibitors in tuberous sclerosis complex. Curr Neuropharmacol 2012; 10: 404–15.
Lasky JL, Wu H . Notch signaling, brain development, and human disease. Pediatr Res 2005; 57: 104R–109R.
Bredel MS, Yadav DM, Alvarez AK, Renfrow AA, Chandler JJ, Yu JP, et al. NFKBIA deletion in glioblastomas. N Engl J Med 2011; 364: 627–37.
Maiese K, Chong ZZ, Shang YC, Wang S . mTOR: on target for novel therapeutic strategies in the nervous system. Trends Mol Med 2013; 19: 51–60.
Kuo YC, Liang CT . Catanionic solid lipid nanoparticles carrying doxorubicin for inhibiting the growth of U87MG cells. Colloids Surf B Biointerfaces 2011; 85: 131–7.
Panja S, Dey G, Bharti R, Kumari K, Maiti TK, Mandal M, et al. Tailor-made temperature-sensitive micelle for targeted and on-demand release of anticancer drugs. ACS Appl Mater Interfaces 2016; 8: 12063–74.
Ziegler DS, Wright RD, Kesari S, Lemieux ME, Tran MA, Jain M, et al. Resistance of human glioblastoma multiforme cells to growth factor inhibitors is overcome by blockade of inhibitor of apoptosis proteins. J Clin Invest 2008; 118: 3109–22.
Roberts WG, Whalen PM, Soderstrom E, Moraski G, Lyssikatos JP, Wang HF, et al. Antiangiogenic and antitumor activity of a selective PDGFR tyrosine kinase inhibitor, CP-673,451. Cancer Res 2005; 65: 957–66.
Polverino A, Coxon A, Starnes C, Diaz Z, DeMelfi T, Wang L, et al. AMG 706, an Oral, Multikinase inhibitor that selectively targets vascular endothelial growth factor, platelet-derived growth factor, and kit receptors, potently inhibits angiogenesis and induces regression in tumor xenografts. Cancer Res 2006; 66: 8715–21.
Stuart DE . NJ 07936 (US), Substituted benzimidazoles for the treatment of astrocytomas. Eur Patent Specification EP 2 391 366 B1, 2012.
Dancey JE . Therapeutic targets: MTOR and related pathways. Cancer Biol Ther 2006; 5: 1065–73.
Lakka SS, Rao JS . Antiangiogenic therapy in brain tumors. Expert Rev Neurother 2008; 8: 1457–73.
Banerjee I, De K, Mukherjee D, Dey G, Chattopadhyay S, Mukherjee M, et al. Paclitaxel-loaded solid lipid nanoparticles modified with Tyr-3-octreotide for enhanced anti-angiogenic and anti-glioma therapy. Acta Biomater 2016; 38: 69–81.
Friedman HS, Kerby T, Calvert H . Temozolomide and treatment of malignant glioma. Clin Cancer Res 2000; 6: 2585–97.
Hart MG, Grant R, Garside R, Rogers G, Somerville M, Stein K . Temozolomide for high grade glioma. Cochrane Database Syst Rev 2013; CD007415.
Olson JJ, Nayak L, Ormond DR, Wen PY . Kalkanis SN ; AANS/CNS Joint Guidelines Committee. The role of cytotoxic chemotherapy in the management of progressive glioblastoma: a systematic review and evidence-based clinical practice guideline. J Neurooncol 2014; 118: 501–55.
Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol 2009; 10: 459–66.
Mrugala MM . Advances and challenges in the treatment of glioblastoma: a clinician's perspective. Discov Med 2013; 15: 221–30.
Chakravarti A, Dicker A, Mehta M . The contribution of epidermal growth factor receptor (EGFR) signaling pathway to radioresistance in human gliomas: a review of preclinical and correlative clinical data. Int J Radiat Oncol Biol Phys 2004; 58: 927–31.
Stommel JM, Kimmelman AC, Ying H, Nabioullin R, Ponugoti AH, Wiedemeyer R, et al. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science 2007; 318: 287–90.
Reardon DA, Rich JN, Friedman HS, Bigner DD . Recent advances in the treatment of malignant astrocytoma. J Clin Oncol 2006; 24: 1253–65.
Capdevila L, Cros S, Ramirez JL, Sanz C, Carrato C, Romeo M, et al. Neoadjuvant cisplatin plus temozolomide versus standard treatment in patients with unresectable glioblastoma or anaplastic astrocytoma: a differential effect of MGMT methylation. J Neurooncol 2014; 117: 77–84.
Mercurio S, Padovani L, Colin C, Carré M, Tchoghandjian A, Scavarda D, et al. Evidence for new targets and synergistic effect of metronomic celecoxib/fluvastatin combination in pilocytic astrocytoma. Acta Neuropathol Commun 2013; 1: 17.
Jozwiak KK . Subependymal giant cell astrocytoma: role of mTOR pathway and its inhibitors. Tumors of the Central Nervous System. 5: 45–55.
Nyfeler B, Chen Y, Li X, Pinzon-Ortiz M, Wang Z, Reddy A, et al. RAD001 enhances the potency of BEZ235 to inhibit mTOR signaling and tumor growth. PLoS One 2012; 7: e48548.
Goudar RK, Shi Q, Hjelmeland MD, Keir ST, McLendon RE, Wikstrand CJ, et al. Combination therapy of inhibitors of epidermal growth factor receptor/vascular endothelial growth factor receptor 2 (AEE788) and the mammalian target of rapamycin (RAD001) offers improved glioblastoma tumor growth inhibition. Mol Cancer Ther 2005; 4: 101–12.
Wang H, Yang Y, Li Y, Bai B, Wang X, Tan H, et al. Systematic optimization of long gradient chromatography mass spectrometry for deep analysis of brain proteome. J Proteome Res 2015; 14: 829–38.
Zhang H, Liu T, Zhang Z, Payne SH, Zhang B, McDermott JE, et al. Integrated proteogenomic characterization of human high-grade serous ovarian cancer. Cell 2016; 166: 755–65.
Li Y, Wang X, Cho JH, Shaw TI, Wu Z, Bai B, et al. JUMPg: an integrative proteogenomics pipeline identifying unannotated proteins in human brain and cancer cells. J Proteome Res 2016; 15: 2309–20
Tobias A, Ahmed A, Moon KS, Lesniak MS . The art of gene therapy for glioma: a review of the challenging road to the bedside. J Neurol Neurosurg Psychiatry 2013; 84: 213–22.
Mizoguchi M, Guan Y, Yoshimoto K, Hata N, Amano T, Nakamizo A, et al. Clinical implications of microRNAs in human glioblastoma. Front Oncol 2013; 3: 19.
Hummel R, Maurer J, Haier J . MicroRNAs in brain tumors : a new diagnostic and therapeutic perspective? Mol Neurobiol 2011; 44: 223–34.
McCord AM, Jamal M, Williams ES, Camphausen K, Tofilon PJ, et al. CD133+ glioblastoma stem-like cells are radiosensitive with a defective DNA damage response compared with established cell lines. Clin Cancer Res 2009; 15: 5145–53.
Clement V, Sanchez P, de Tribolet N, Radovanovic I, Ruiz i Altaba A, et al. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr Biol 2007; 17: 165–72.
Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B, et al. A perivascular niche for brain tumor stem cells. Cancer Cell 2007; 11: 69–82.
Chow KH, Gottschalk S . Cellular immunotherapy for high-grade glioma. Immunotherapy 2011; 3: 423–34.
Mitchell DA, Sampson JH . Toward effective immunotherapy for the treatment of malignant brain tumors. Neurotherapeutics 2009; 6: 527–38.
Okada H, Kohanbash G, Zhu X, Kastenhuber ER, Hoji A, Ueda R, et al. Immunotherapeutic approaches for glioma. Crit Rev Immunol 2009; 29: 1–42.
Johnson LA, Sampson JH . Immunotherapy approaches for malignant glioma from 2007 to 2009. Curr Neurol Neurosci Rep 2010; 10: 259–66.
Yu JS, Liu G, Ying H, Yong WH, Black KL, Wheeler CJ, et al. Vaccination with tumor lysate-pulsed dendritic cells elicits antigen-specific, cytotoxic T-cells in patients with malignant glioma. Cancer Res 2004; 64: 4973–9.
Pellegatta S, Poliani PL, Corno D, Menghi F, Ghielmetti F, Suarez-Merino B, et al. Neurospheres enriched in cancer stem-like cells are highly effective in eliciting a dendritic cell-mediated immune response against malignant gliomas. Cancer Res 2006; 66: 10247–52.
Schmitz M, Temme A, Senner V, Ebner R, Schwind S, Stevanovic S, et al. Identification of SOX2 as a novel glioma-associated antigen and potential target for T cell-based immunotherapy. Br J Cancer 2007; 96: 1293–301.
Rosenberg SA . Cell transfer immunotherapy for metastatic solid cancer — what clinicians need to know. Nat Rev Clin Oncol 2011; 8: 577–85.
Cheadle EJ, Sheard V, Hombach AA, Chmielewski M, Riet T, Berrevoets C, et al. Chimeric antigen receptors for T-cell based therapy. Methods Mol Biol 2012; 907: 645–66.
Lamers CH, Sleijfer S, Vulto AG, Kruit WH, Kliffen M, Debets R, et al. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol 2006; 24: 20–2.
Till BG, Jensen MC, Wang J, Qian X, Gopal AK, Maloney DG, et al. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: pilot clinical trial results. Blood 2012; 119: 3940–50.
Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med 2008; 14: 1264–70.
Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med 2013; 5: 177ra38.
Porter DL, Levine BL, Kalos M, Bagg A, June CH, et al. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med 2011; 365: 725–33.
Brentjens R, Yeh R, Bernal Y, Riviere I, Sadelain M, et al. Treatment of chronic lymphocytic leukemia with genetically targeted autologous T cells: case report of an unforeseen adverse event in a phase I clinical trial. Mol Ther 2010; 18: 666–8.
Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood 2012; 119: 2709–20.
Moscatello DK, Holgado-Madruga M, Godwin AK, Ramirez G, Gunn G, Zoltick PW, et al. Frequent expression of a mutant epidermal growth factor receptor in multiple human tumors. Cancer Res 1995; 55: 5536–9.
Miao H, Choi BD, Suryadevara CM, Sanchez-Perez L, Yang S, De Leon G, et al. EGFRvIII-specific chimeric antigen receptor T cells migrate to and kill tumor deposits infiltrating the brain parenchyma in an invasive xenograft model of glioblastoma. PLoS One 2014; 9: e94281.
Grossauer S, Koeck K, Petritsch C . Immune checkpoint blockage — a promising strategy to overcome glioma stem cell therapy-resistance. Insights Neurosurgery 2015; 1: 1–8.
Kim ES, Kim JE, Patel MA, Mangraviti A, Ruzevick J, Lim M, et al. Immune checkpoint modulators: an emerging antiglioma armamentarium. J Immunol Res 2016; 2016: 4683607.
Fecci PE, Ochiai H, Mitchell DA, Grossi PM, Sweeney AE, Archer GE, et al. Systemic CTLA-4 blockade ameliorates glioma-induced changes to the CD4+ T cell compartment without affecting regulatory T-cell function. Clin Cancer Res 2007; 13: 2158–67.
Huang BY, Zhan YP, Zong WJ, Yu CJ, Li JF, Qu YM, Han S, et al. The PD-1/B7-H1 pathway modulates the natural killer cells versus mouse glioma stem cells. PLoS One 2015; 10: 0134715.
Wainwright DA, Chang AL, Dey M, Balyasnikova IV, Kim CK, Tobias A, et al. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clin Cancer Res 2014; 20: 5290–301.
Belcaid Z, Phallen JA, Zeng J, See AP, Mathios D, Gottschalk C, et al. Focal radiation therapy combined with 4-1BB activation and CTLA-4 blockade yields long-term survival and a protective antigen specific memory response in a murine glioma model. PLoS One 2014; 9.
Kim JE PM, Mangraviti A, Velarde E, Theodros D, Mathios D, et al. The combination of anti-TIM-3 and anti-PD-1 checkpoint inhibitors with focused radiation resulted in a synergistic antitumor immune response in a preclinical glioma model. Neurosurgery 2015; 1: 212.
Nicholas S, Mathios D, Ruzevick J, Jackson C, Yang I, Lim M, et al. Current trends in glioblastoma multiforme treatment: radiation therapy and immune checkpoint inhibitors. Brain Tumor Res Treat 2013; 1: 2–8.
Heimberger AB, Sun W, Hussain SF, Dey M, Crutcher L, Aldape K, et al. Immunological responses in a patient with glioblastoma multiforme treated with sequential courses of temozolomide and immunotherapy: case study. Neuro Oncol 2008; 10: 98–103.
Banissi C, Ghiringhelli F, Chen L, Carpentier AF . Treg depletion with a low-dose metronomic temozolomide regimen in a rat glioma model. Cancer Immunol Immunother 2009; 58: 1627–34.
Cockle JV, Rajani K, Zaidi S, Kottke T, Thompson J, Diaz RM, et al. Combination viroimmunotherapy with checkpoint inhibition to treat glioma, based on location-specific tumor profiling. Neuro Oncol 2016; 18: 518–27.
Acknowledgements
We would like to thank Dr Sanjoy CHATTERJEE and Dr Rimpa Basu ACHARI (Tata Medical Center, Kolkata, India) for their advice and comments on the paper. The authors are thankful to DST–INSPIRE (IF130658), DBT, CSIR-UGC and BRNS, India for providing research fellowships.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary files are available at the website of Acta Pharmacologica Sinica.
Rights and permissions
About this article

Cite this article
Rajesh, Y., Pal, I., Banik, P. et al. Insights into molecular therapy of glioma: current challenges and next generation blueprint. Acta Pharmacol Sin 38, 591–613 (2017). https://doi.org/10.1038/aps.2016.167
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2016.167
Keywords
This article is cited by
-
MZ1, a BRD4 inhibitor, exerted its anti-cancer effects by suppressing SDC1 in glioblastoma
BMC Cancer (2024)
-
The genetic liability to rheumatoid arthritis may decrease hepatocellular carcinoma risk in East Asian population: a Mendelian randomization study
Arthritis Research & Therapy (2023)
-
The alternative splicing of intersectin 1 regulated by PTBP1 promotes human glioma progression
Cell Death & Disease (2022)
-
Circular RNA circ_0079593 facilitates glioma development via modulating miR-324-5p/XBP1 axis
Metabolic Brain Disease (2022)
-
Pediatric primary high-grade spinal glioma: a National Cancer Database analysis of current patterns in treatment and outcomes
Child's Nervous System (2021)
