
Abstract
We previously identified AG-690/11026014 (6014) as a novel poly(ADP-ribose) polymerase-1 (PARP-1) inhibitor that effectively prevented angiotensin II (Ang II)-induced cardiomyocyte hypertrophy. In the present study, we reported a new synthesis route for 6014, and investigated its protective effects on Ang II-induced cardiac remodeling and cardiac dysfunction and the underlying mechanisms in mice. We designed a new synthesis route to obtain a sufficient quantity of 6014 for this in vivo study. C57BL/6J mice were infused with Ang II and treated with 6014 (10, 30, 90 mg·kg−1·d−1, ig) for 4 weeks. Then two-dimensional echocardiography was performed to assess the cardiac function and structure. Histological changes of the hearts were examined with HE staining and Masson's trichrome staining. The protein expression was evaluated by Western blot, immunohistochemistry and immunofluorescence assays. The activities of sirtuin-1 (SIRT-1) and the content of NAD+ were detected with the corresponding test kits. Treatment with 6014 dose-dependently improved cardiac function, including LVEF, CO and SV and reversed the changes of cardiac structure in Ang II-infused mice: it significantly ameliorated Ang II-induced cardiac hypertrophy evidenced by attenuating the enlargement of cardiomyocytes, decreased HW/BW and LVW/BW, and decreased expression of hypertrophic markers ANF, BNP and β-MHC; it also prevented Ang II-induced cardiac fibrosis, as implied by the decrease in excess accumulation of extracellular matrix (ECM) components collagen I, collagen III and FN. Further studies revealed that treatment with 6014 did not affect the expression levels of PARP-1, but dose-dependently inhibited the activity of PARP-1 and subsequently restored the activity of SIRT-1 in heart tissues due to the decreased consumption of NAD+ and attenuated Poly-ADP-ribosylation (PARylation) of SIRT-1. In conclusion, the novel PARP-1 inhibitor 6014 effectively protects mice against AngII-induced cardiac remodeling and improves cardiac function. Thus, 6014 might be a potential therapeutic agent for heart diseases..
Similar content being viewed by others


Introduction
Cardiac remodeling, manifested clinically as hypertrophy (enlargement), fibrosis, and dysfunction of the heart resulting from cardiac load or injury, is generally accepted as a determinant of the pathogenesis of heart failure (HF)1. The renin-angiotensin-aldosterone system (RAAS) plays a pivotal role in the physiological and pathological processes of cardiac remodeling. It is also widely accepted that angiotensin II (Ang II), an important component of the RAAS, could induce cardiac remodeling following sustained hypertension2,3,4,5,6.
PARylation, induced by poly(ADP-ribose) polymerases (PARPs), is a type of reversible posttranslational modification impacting numerous cellular processes, eg, DNA repair, transcription, metabolism, or immune functions7. PARP-1, the predominant member of the PARPs, accounts for approximately 90% of the cellular PARylation events8. Once activated, PARP-1 catalyzes protein PARylation, which results in the formation of polymerized ADP-ribose (PAR) dependently on NAD+ donor molecules, and subsequently the covalent attachment of PAR polymers to target proteins9.
PARP-1 is involved in pathological cardiac remodeling and heart failure. Inhibition of PARP-1 prevents pathological cardiac remodeling induced by partial abdominal aortic constriction10. PARP-1-deficient mice are protected from Ang II-induced cardiac hypertrophy11. Moreover, PARP-1 activation facilitates the pathogenesis of cardiac fibrosis12 and the development of heart failure13,14. These findings indicate that PARP-1 could serve as a promising target for cardiac diseases, and the pharmacologic inhibition of PARP-1 may be a potential therapeutic strategy15,16,17.
A previous study from our laboratory identified AG-690/ 11026014(6014) as a novel PARP-1 inhibitor from the Specs database (www.specs.net), which contains approximately 200,000 compounds based on virtual screening. We also reported that 6014 could significantly attenuate Ang II-induced cardiomyocyte hypertrophy18. To further determine the in vivo cardiac protective effect of 6014, the present study used C57BL/6J mice infused with Ang II as the model, and elucidated the effects and underlying mechanisms of this novel PARP-1 inhibitor. This study aimed to determine whether 6014, as a PARP-1 inhibitor, has therapeutic potential for cardiac remodeling.
Materials and methods
Reagents
The compounds 3-aminobenzamide (3-AB), 4',6-diamidino-2-phenylindole (DAPI) and Ang II were purchased from Sigma-Aldrich (St Louis, MO, USA). Anti-PARP-1, anti-collagen I and anti-collagen III antibodies were provided by Proteintech Group (Chicago, IL, USA). Anti-ANF and anti-FN antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-BNP antibody was obtained from Merck Millipore (Bedford, Ohio, USA). Anti-PAR antibody was obtained by Trevigen (Gaithersburg, MD, USA). Anti-SIRT-1, anti-lamin B1, anti-GAPDH antibodies and all secondary antibodies for Western blot experiments were purchased from Cell Signaling Technology (Boston, MA, USA). An hematoxylin and eosin (HE) staining kit was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). A Masson's staining kit and immunohistochemistry reagents including Cy3-conjugated goat anti-mouse IgG (H+L) were purchased from Wuhan Goodbio Technology Co, Ltd (Wuhan, China). RIPA lysis buffer and IP lysis buffer were purchased from Beyotime Biotechnology (Jiangsu, China). The protease and phosphatase inhibitor mini tablets, BCA protein assay kit, Bradford protein assay kit, and protein G-agarose beads were obtained from Pierce Biotechnology (Rockford, AL, USA). A nuclear extract kit was purchased from ACTIVE MOTIF (Carlsbad, NM, USA). The quick start Bradford dye reagent was obtained from Bio-Rad (Richmond, VA, USA). A SIRT-1 deacetylase fluorometric assay kit was purchased from CycLex Ltd (Nagano, Japan).
The inhibitor 6014 was synthesized in our lab. The purities of 6014 used for biological evaluation (99.61%) were determined on a DIONEX Ultimate 3000 HPLC system (Chromeleon SR9 Build 2673): column, Acclaim 120 C18, 5 mm, 4.6 mm×250 mm; flow rate, 1 mL/min. The samples were eluted with a gradient of 5% to 95% solvent A for 25 min and detected at 254 nm; solvent A was CH3CN in double-deionized H2O. 1H NMR and 13C NMR spectra (AvanceIII, Burker) were recorded using TMS as an internal standard with a BurkerBioSpinUltrashield 400 NMR system. High-resolution mass spectra (HRMS) (LCMS-IT-TOF, Shimadzu) were recorded on a Shimadzu LCMS-IT-TOF Instrument.
General procedure for the synthesis of 6014
(1) 4-chlorophenol (77.8 mmol), 2-(chloromethyl)oxirane (306.9 mmol) and K2CO3 (231.5 mmol) were refluxed in acetone (100 mL) overnight, cooled to room temperature and filtered. The filtrate was concentrated and extracted with toluene and water, and then the 2-((4-chlorophenoxy)methyl)oxirane (M1) in toluene was purified by column chromatography.
(2) 3-methyl-1H-purine-2,6(3H,7H)-dione (30 mmol) and AcONa (60 mmol) were dissolved in AcOH, and Br2 was added dropwise and reacted for 2 h at 65 °C. 8-Bromo-3-methyl-1H-purine-2,6(3H,7H)-dione (M2) was generated by cooling, filtering, water washing and drying in a vacuum.
(3) M1 (31.0 mmol) and M2 (25.8 mmol) in ethanol (120 mL) were combined with trimethylamine (2.6 mmol) and refluxed for 3 h. 8-Bromo-7-(3-(4-chlorophenoxy)-2-hydroxypropyl)-3-methyl-1H-purine-2,6(3H,7H)-dione (M3) was produced by cooling, filtering, ethanol washing and drying in a vacuum.
(4) M3 (8.15 mmol) and 2-mercaptoacetic acid (16.3 mmol) in DMF (35 mL) were combined with NaHCO3 (32.6 mmol) and reacted for 2 h at 85 °C. The reaction was stopped with water and then washed by methyl tertiary butyl ether. The products in the water were combined with HCl (2.0 mmol) until the pH reached 3, and 2-((7-(3-(4-chlorophenoxy)-2-hydroxypropyl)-3-methyl-2,6-dioxo-2,3,6,7-tetrahydro-1H-purin-8-yl)thio)acetic acid (M4) was yielded after filtering, water washing and drying in a vacuum.
(5) M4 (7.26 mmol) and ammonium chloride (21.7 mmol) in DMF (40 mL) was added to HATU (8.71 mmol) and DIEA (21.8 mmol) on ice and reacted overnight at room temperature. The products were precipitated by adding water and then filtering. 2-((7-(3-(4-chlorophenoxy)-2-hydroxypropyl)-3-methyl-2,6-dioxo-2,3,6,7-tetrahydro-1H-purin-8-yl)thio)acetamide (6014) was purified by preparative chromatography (Varian, Agilent).
The synthesized intermediates M1-M4 were structurally identified by 1H NMR or MS, and 6014 was structurally identified by 1H NMR, 13C NMR and HRMS.
2-((4-chlorophenoxy)methyl)oxirane (M1)
Crystal oil, yield 62.0%; 1H NMR (400 MHz, CDCl3) δ 7.28–7.22 (m, 2H), 6.90–6.83 (m, 2H), 4.23 (dd, J=11.0, 3.0 Hz, 1H), 3.93 (dd, J=11.0, 5.7 Hz, 1H), 3.39–3.32 (m, 1H), 2.92 (t, J=4.5 Hz, 1H), 2.76 (dd, J=4.8, 2.6 Hz, 1H).
8-bromo-3-methyl-1H-purine-2,6(3H,7H)-dione (M2)
White solid, yield 95%; 1H NMR (400 MHz, DMSO) δ 14.29 (s, 1H), 11.18 (s, 1H), 3.32 (s, 3H). MS (ESI, neg ion) m/z: 242.9 [M-H]−
8-bromo-7-(3-(4-chlorophenoxy)-2-hydroxypropyl)-3-methyl-1H-purine-2,6(3H,7H)-dione (M3)
White solid, yield 85.9%; MS (ESI, neg ion) m/z: 427.0 [M-H]−
2-((7-(3-(4-chlorophenoxy)-2-hydroxypropyl)-3-methyl-2,6-dioxo-2,3,6,7-tetrahydro-1H-purin-8-yl)thio)acetic acid (M4)
White solid, yield 90.5%; MS (ESI, neg ion) m/z: 439.0 [M-H]−
2-((7-(3-(4-chlorophenoxy)-2-hydroxypropyl)-3-methyl-2,6-dioxo-2,3,6,7-tetrahydro-1H-purin-8-yl)thio)acetamide (6014)
White solid, yield 30%; HPLC tR=11.24 min; 1H NMR (400 MHz, DMSO) δ 11.11 (s, 1H), 7.65 (s, 1H), 7.32 (d, J=8.7 Hz, 2H), 7.23 (s, 1H), 6.93 (d, J=8.7 Hz, 2H), 5.52 (d, J=4.9 Hz, 1H), 4.37 (d, J=9.7 Hz, 1H), 4.27–4.15 (m, 2H), 4.01 (dd, J=9.9, 3.3 Hz, 1H), 3.98–3.91 (m, 3H). 13C NMR (101 MHz, DMSO) δ 168.66, 157.24, 153.93, 150.60, 150.53, 149.58, 129.14, 124.36, 116.26, 108.11, 70.33, 67.65, 48.91, 36.68, 28.44. HRMS calcd. for C17H18N5O5SCl [M+H]+: 440.0790, found 440.0763.
Animals
Healthy adult male C57BL/6J mice (10 weeks old, 20–25 g weight) were raised in the animal center of Sun Yat-Sen University and kept under pathogen-free conditions at approximately 22 °C. All experimental procedures were conducted in accordance with the Guidelines of Animal Experiments from the Ethical Committee for Animal Research of Sun Yat-sen University (No 44008500011624).
Minipump implantation and administration
As a commonly used PARP-1 inhibitor19,20, 3-aminobenzamide (3-AB) was used as a positive control in this study. The C57BL/6J male mice were randomly assigned to 6 groups, n=12/group: ① sham group; ② Ang II group; ③ Ang II+3-AB (30); ④ Ang II+6014 (10); ⑤ Ang II+6014 (30); ⑥ Ang II+6014 (90). 3-AB (30), 3-AB (30 mg·kg−1·d−1); 6014 (10), 6014 (10 mg·kg−1·d−1); 6014 (30), 6014 (30 mg·kg−1·d−1); 6014 (90), 6014 (90 mg·kg−1·d−1).
Mice from all groups except the sham group were infused with Ang II (1000 ng·kg−1·min−1) via subcutaneously implanted minipumps (Alzet® pump model 2004, Cupertino, USA) for 4 weeks. Control mice were infused with normal saline. After the minipumps were implanted, administration of 3-AB and 6014 were carried out by intragastric administration twice a day, and both 3-AB and 6014 were suspended in sodium carboxymethylcellulose.
Echocardiography
At the end of the experiment, all the mice were anesthetized with 1.5% isoflurane in oxygen (1 L/min). Two-dimensionally guided M-mode echocardiography (ESAOTE, Italy) was performed using a Technos MPX ultrasound system equipped with a 21-MHz imaging transducer, as described previously21. Ultrasound images of the heart were obtained by gently applying the probe to the chest. The M-mode images were obtained from the parasternal short-axis view at the papillary muscle. The following parameters were measured and calculated: cardiac output (CO), left ventricular fractional shortening (LVFS), left ventricular ejection fraction (LVEF), heart rate (HR), stroke volume (SV), left ventricular internal dimension (LVID) in diastole and systole (LVIDd and LVIDs, respectively), left ventricular posterior wall (LVPW) thickness in diastole and systole (LVPWd and LVPWs, respectively), and the interventricular septum (IVS) dimension in diastole and systole (IVSd and IVSs, respectively). All the parameters were obtained from three cardiac cycles, and the data were analyzed by GraphPad Prism 5 (GraphPad Inc, La Jolla, CA, USA).
Collection of tissues
After the echocardiography measurements were completed, all the mice were weighed and anesthetized by sodium pentobarbital (40 mg/kg). The hearts of the mice were rapidly removed, imaged by photography, and weighed. Then, the left ventricle was carefully trimmed and weighed. The ratio of heart weight to body weight (heart weight/body weight, HW/BW) and left ventricular weight to body weight (left ventricular weight/body weight, LVW/BW) were analyzed.
Half of the hearts from each group were stored in formalin and then embedded in 4% paraformaldehyde for later histopathological analysis. For the other half, the left ventricles were isolated and weighed. The left ventricles of the heart were snap frozen in liquid nitrogen for Western blot analysis, SIRT-1 activity measurement and NAD+ content detection.
Histopathological analysis
For immunohistochemical staining, the tissues were fixed in 4% formalin and embedded in paraffin. Sections (5 mm) were cut from the embedded blocks and floated onto a warm (42 °C) water bath from where they were placed on Superfrost plus slides. The sections were deparaffinized and hydrated, and the endogenous peroxidase activity was blocked with 3% H2O2 in water for 10 min. Antigen retrieval was performed with 10 mmol/L citrate buffer (pH 6.0) for 10 min. The slides were incubated with blocking reagent (Thermo Scientific) for 10 min to block nonspecific binding. Then, they were incubated with anti-PAR antibodies overnight at 4 °C, and later with HRP-conjugated secondary antibodies for 30 min at room temperature. Finally, the slides were incubated with diaminobenzidine and counterstained with HE.
For HE staining, after deparaffinization and hydration, the sections were stained with HE. For Masson's staining, after deparaffinization and hydration, the sections were stained with hematoxylin and Ponceau, as well as with phosphomolybdic acid and aniline blue successively in Masson's staining.
For immunofluorescence, the frozen sections were fixed by acetone. Bovine serum albumin (BSA) was used to block non-specific binding after antigen retrieval. The tissue sections were incubated with anti-PAR antibody (1:100) overnight at 4 °C and incubated with Cy3-conjugated goat anti-mouse IgG (H+L) for 1 h. Subsequently, the nucleus was stained with DAPI. All the stained sections were sealed with neutral resin and photographed under an inverted fluorescence microscope (Olympus, Japan).
Western blotting assay
To extract the total protein, the mouse left ventricular tissue was homogenized in RIPA lysis buffer containing the protease inhibitor PMSF, and the total protein contents were determined with a BCA protein assay kit (Pierce). To extract the nucleoprotein, a nuclear extract kit (ACTIVE MOTIF) was used according to the manufacturer's instructions, and the nucleoprotein contents were determined with a Bradford protein assay kit (Pierce). Then, equal amounts of protein samples were separated by SDS-PAGE and then transferred to PVDF membranes (Millipore, MA, USA). After blocking with 5% (w/v) nonfat milk (Bio-Rad) dissolved in Tris-Tween 20 (TBS-T) for 1 h at room temperature, the membranes were incubated with anti-GAPDH, anti-lamin B1, anti-ANF, anti-BNF, anti-β-MHC, anti-collagen I, anti-collagen III, anti-FN, anti-PARP-1, anti-SIRT-1 or anti-PAR antibodies overnight at 4 °C. Then, horseradish peroxidase (HRP)-labeled secondary antibodies were added for incubation for 1 h at room temperature. Immunoreactive bands were visualized with SuperSignal West Pico Chemiluminescent Substrate (Pierce). The intensity of target protein bands was quantified with Image J software (National Institutes of Health, USA) and normalized to GAPDH or Lamin B1 as loading control.
Co-Immunoprecipitation
To investigate whether there was an interaction between SIRT-1 and PARP-1, a co-immunoprecipitation (co-IP) assay was performed. Nuclear proteins (800 μg) were incubated with 3 μg anti-SIRT-1 or anti-PARP-1 antibodies overnight (rabbit normal IgG was used as a control), followed by a 4 h incubation with protein G-agarose beads (Pierce, Rockford, IL, USA) at 4 °C. The immunoprecipitated proteins were detected by Western blot.
Detection of SIRT-1 activity
To detect the activity of SIRT-1, a SIRT-1 deacetylase fluorometric assay kit (CycLex Ltd) was used. In this method, the reaction was initiated and the fluorescence intensity was measured by mixing simultaneously fluorescence-labeled acetylated peptide (substrate), NAD+, and developer with SIRT-1 samples. SIRT-1 samples were obtained in the immunoprecipitation assays using anti-SIRT-1 antibody. The fluorescence intensity was measured at 490±10 nm (excitation) and 530±10 nm (emitted light) by an automatic microplate reader (Tecan, Switzerland).
Determination of NAD+ content
The NAD+ levels were measured according to a method described previously22. Briefly, the frozen crushed tissue was suspended in perchloric acid before centrifugation. The supernatant was neutralized with KOH and KH2PO4/K2HPO4, and then added to the NAD+ reaction mixture containing ethanol, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, phenazine ethosulfate, EDTA, BSA, and bicine. The reaction was initiated by alcohol dehydrogenase and stopped by iodoacetate after incubation for 20 min at 37 °C. The absorbance of the reaction mixture was read at a wavelength of 570 nm. The NAD+ content was calculated from the standard curve and normalized to the protein content of the sample.
Statistical analysis
Statistical analysis data are presented as the mean±SEM of at least three independent experiments. Statistical analysis was performed using a two-tailed unpaired Student's t-test between two groups and by one-way ANOVA followed by Bonferroni's test for multiple comparisons. The analyses were performed using GraphPad Prism 5.0 (GraphPad Inc, La Jolla, CA, USA). P<0.05 was considered statistically significant.
Results
Synthesis of 6014
To obtain a sufficient quantity of 6014 for the in vivo study, a novel synthetic route of 6014 was designed that is illustrated in Scheme 1. In brief, 6014 was synthesized through five steps, three of which could be completed simply and independently on a column chromatograph to obtain pure product. This efficient synthetic route of 6014 was reported for the first time herein, and could be worthy for large scale synthesis. Finally, an adequate amount of 6014 (2.0 g) was obtained and successfully used for animal experiments. The purity and structure of 6014 were confirmed by 1H NMR, 13C NMR, HRMS and HPLC (Figure S1, S2, S3, S4).

Synthetic route of AG-690/11026014.
6014 prevented cardiac dysfunction and changes of cardiac structure in Ang II-infused mice
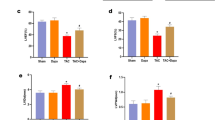
To investigate the protective effect of 6014 on cardiac function and structure, two-dimensional echocardiography was performed (Table S1). The echocardiography results indicated that the LVPWd, LVPWs, IVSd, IVSs were remarkably increased, while the LVIDd and LVIDs were decreased in Ang II-infused mice. The inhibitor 6014 dose-dependently reversed these Ang II-induced changes, similarly as the well-known PARP-1 inhibitor 3-AB (Figure 1A–1G). The LVEF, LVFS, SV and CO have been most widely used to evaluate left ventricular systolic function. Uniquely, LVFS was not significantly different among mice receiving different treatments (Table S1). Treatment with 6014 attenuated the Ang II-induced increases of LVEF, SV and CO in a dose-dependent manner (Figure 1H–1J).

6014 reversed the cardiac dysfunction and changes of cardiac structure induced by Ang II. Representative echocardiographic graphs (A) were shown and IVSd (B), LVPWd (C), LVIDd (D), IVSs (E), LVPWs (F), LVIDs (G), LVEF (H), SV (I), and CO (J) were measured and calculated from parameters of echocardiography. Data were expressed as mean±SEM from three independent experiments. **P<0.01 vs Sham. ##P<0.01 vs Ang II.
6014 attenuated cardiac hypertrophy and fibrosis induced by Ang II
As shown in Figure 2A, Ang II treatment led to the enlargement of heart size, which could be prevented by 6014 at the doses of 30 and 90 mg·kg−1·d−1, as well as 3-AB at a dose of 30 mg·kg−1·d−1. Statistical results demonstrated that the ratios of HW/BW and LVW/BW were increased to 6.38±0.55 mg/g and 3.70±0.43 mg/g respectively by stimulation with Ang II, while 6014 (and 10, 30 and 90 mg·kg−1·d−1) dose-dependently inhibited the increase of HW/BW (5.97±0.55, 5.72±0.53 and 5.46±0.61 mg/g) and LVW/BW (3.65±0.42, 3.26±0.23 and 3.09±0.26 mg/g) (Figure 2D and 2E). HE staining images also showed that 6014 significantly prevented the enlargement of cardiomyocytes induced by Ang II (Figure 2B).

6014 attenuated the enlargement of the heart size, cardiomyocytes size and prevented the accumulation of collagen. (A) Representative diagram of mice hearts under different treatment. (B) Representative images diagram of H&E staining of cardiomyocytes. (C) Representative images of Masson's staining of total collagen in tissue sections of heart. (D) Statistical results of HW/BW. (E) Statistical results of LVW/BW. (F) Statistical areas of total collagen production versus areas of whole tissue. Data were expressed as mean±SEM from three independent experiments. **P<0.01 vs Sham. #P<0.05, ##P<0.01 vs Ang II.
Additionally, a significant increase of total collagen in the hearts of the Ang II group was observed by the Masson's staining assays, which represented the appearance of cardiac fibrosis (Figure 2C). Treatment with 6014 remarkably and dose-dependently prevented the accumulation of collagen (10.3%±1.5%, 4.2%±0.76% and 3.0%±0.15%) compared to the Ang II group (14.4%±2.0%) (Figure 2C and 2F). The effect of 6014 was comparable to that of 3-AB (6.4%±0.85%) (Figure 2C and 2F).
Western blot analysis showed that 6014 dose-dependently alleviated the expression of the hypertrophic marker proteins (ANF, BNP and β-MHC) (Figure 3A–3C) and the fibrosis marker proteins (collagen I, collagen III and FN) (Figure 3D–3F), in contrast to Ang II. Taken together, these results suggested that 6014 could markedly prevent cardiac hypertrophy and fibrosis induced by Ang II.

Effects of 6014 on the expression of hypertrophy marker proteins and fibrosis marker proteins. Protein expression of ANF (A), BNP (B), β-MHC (C), FN (D), collagen I (E) and collagen III (F) were measured by Western blot. Data were expressed as mean±SEM from duplicates in three independent experiments. **P<0.01 vs Sham. #P<0.05, ##P<0.01 vs Ang II.
Effects of 6014 on the expression and enzymatic activity of PARP-1
PARP-1 localizes mainly to the cell nucleus and to a lesser extent in the mitochondria23,24,25; therefore, the expression of nuclear and total PARP-1 was detected by Western blot. As shown in Figure 4A, 4B, both the nuclear and total expression of PARP-1 was up-regulated upon stimulation of Ang II but was unaltered by 3-AB and 6014. Target proteins undergo the PARylation modification when PARP-1 is activated. Thus, the enzymatic activity of PARP-1 was evaluated by the expression of PARylated proteins using anti-PAR antibody. As shown in Figure 4C, Ang II clearly stimulated the PARylation of proteins (3.20±0.30), whereas 6014 dose-dependently inhibited the increased expression of PARylated proteins (3.10±0.26, 1.53±0.06 and 1.20±0.10).

Effects of 6014 on the expression and enzymatic activity of PARP-1. The expression of PARP-1 in nucleus (A) and whole cell (B) as well as PARylated proteins in whole cell (C). Data were expressed as mean±SEM from duplicates in three independent experiments. *P<0.05, **P<0.01 vs Sham. ##P<0.01 vs Ang II.
Immunohistochemistry and immunofluorescence assays were subsequently performed to further confirm the inhibition of 6014 on PARP-1 activity. Representative images of immunohistochemistry are shown in Figure 5A, 5B. Ang II clearly increased the expression of PARylated proteins, which were labeled with the anti-PAR antibody (brown). The up-regulation of PARylated proteins was prominently inhibited by the treatment of 6014 at the doses of 30 and 90 mg·kg−1·d−1, comparable to 3-AB (30 mg·kg−1·d−1). Likewise, the fluorescence intensity of PARylated proteins (red) was augmented in the hearts of Ang II-infused mice but was suppressed by 6014 (30 and 90 mg·kg−1·d−1) and 3-AB (30 mg·kg−1·d−1) (Figure 5C). Therefore, these observations were in line with the Western blot results (Figure 4C), confirming that 6014 efficiently inhibited the activation of PARP-1 but did not affect its protein expression in Ang II-infused mice.

Effects of 6014 on enzymatic activity of PARP-1. (A, B) Representative images of PARylated proteins expression by immunohistochemistry. PARylated proteins were stained with anti-PAR antibody (brown). (C) Representative images of PARylated proteins by immunofluorescence. PARylated proteins were stained with anti-PAR antibody (red) and nuclei were stained with DAPI (blue).
6014 inhibited PARylation of SIRT-1 and restored the activity of SIRT-1 in Ang II-infused mice
Ang II treatment significantly decreased the NAD+ content (80.3%±4.5%) and SIRT-1 activity (42.7%±5.0%) in left ventricles (Figure 6A and 6B). Treatment with 6014 did not retard the decrease of NAD+ at 10 and 30 mg·kg−1·d−1 but did restore the NAD+ content to 95.3%±5.0% of the sham group at the high dose of 90 mg·kg−1·d−1 (Figure 6A), which may due to the strong inhibition of PARP-1 by 6014. SIRT-1 is an NAD+ dependent enzyme that can compete with PARP-1 in NAD+ consumption. Interestingly, 6014 could strongly restore SIRT-1 activity at the medium dose of 30 mg·kg−1·d−1 (66.3%±9.1%) compared to the Ang II (42.7%±5.0%), even though the NAD+ content was not increased by 6014 at the same dose (Figure 6A and 6B). The results indicated that other factors underlying the SIRT-1 activity regulation may exist in addition to the restoration of NAD+content by 6014.

Effects of 6014 on the regulation and activity of SIRT-1. NAD+ content analysis (A) and SIRT-1 activity (B) were analyzed. Protein expression of SIRT-1 were measured by Western blot (C). The interaction between SIRT-1 and PARP-1 (D) and the PARylation of SIRT-1 (E) were measured by co-IP assay. Data were expressed as mean±SEM from three independent experiments. *P<0.05, **P<0.01 vs Sham. #P<0.05, ##P<0.01 vs Ang II.
Western blot analysis showed that 6014 dose-dependently alleviated the expression of SIRT-1 in contrast to Ang II (Figure 6C), and the trend was consistent with that of SIRT-1 activity. SIRT-1 was precipitated by anti-PARP-1 (Figure 6D), suggesting an interaction between SIRT-1 and PARP-1. Additionally, Ang II enhanced this interaction significantly, and 6014 attenuated the enhancement (Figure 6D). Moreover, the PARylation of SIRT-1 was detected using an anti-PAR antibody (Figure 6E). We observed two bands (approximately 170 and 250 kDa), which were present at higher levels than the SIRT-1 band (120 kDa) (Figure 6E), suggesting the PARylation of SIRT-1. The results indicated 6014 significantly reduced the PARylation of SIRT-1, in contrast to Ang II, which might contribute to its activation of SIRT-1 activity.
Discussion
In this study, we first designed a novel route of synthesis and successfully synthesized a sufficient amount of 6014. During cardiac remodeling, the most apparent change is the enlargement of the heart, i.e., cardiac hypertrophy. In the cardiac remodeling mouse model induced by Ang II, we observed that 6014 ameliorated cardiac hypertrophy by attenuating the enlargement of cardiomyocytes and the increase of HW/BW and LVW/BW and by decreasing the expression of fetal genes, including ANF, BNP and β-MHC. Cardiac hypertrophy, only when accompanied by fibrosis during remodeling, can lead to cardiac dysfunction26. It was also found that PARylation of PARP-1 is responsible for the pathogenesis of tissue fibrosis27,28. Some researchers have reported that PARylation of c-Jun/c-Fos makes an important contribution to Ang II-induced cardiac fibrosis29, and the inhibition of PARP-1 prevented Ang II-induced aortic fibrosis in rats30. Herein, we also found that 6014 prevented cardiac fibrosis induced by Ang II, as implied by the decrease of excess accumulation of extracellular matrix (ECM) components, including collagen I, collagen III and FN. Moreover, 6014 improved cardiac function, including LVEF, CO and SV and reversed the changes of cardiac structure in Ang II-infused mice. Above all, these results confirmed for the first time that 6014 exerts cardiac protective effects against cardiac remodeling. Importantly, 6014 showed better cardiac protective effects and lower cytotoxicity (Figure S5) than 3-AB, which proved it to be very meaningful to investigate its cardio-protection in vivo.
Our previous work identified 6014 as a novel PARP-1 inhibitor with obvious inhibition of the activity of PARP-1 at the enzyme level and in a cardiomyocyte model. However, the underlying mechanisms of its cardiac protective effects in vivo remained unexplored. Herein, we further demonstrated that the cardio-protective effects of 6014 depended on the inhibition of PARylation, which was shown to activate SIRT-1.
SIRT-1, a prototypical member of the sirtuin family, is an NAD+-dependent deacetylase enzyme31. Recently, SIRT-1 has gathered significant interest due to its cardiovascular protective role32,33. Several studies focusing on the responses of resveratrol (RES) as an accepted SIRT-1 activator also indicated that SIRT-1 may have a beneficial role in heart diseases and that the up-regulation of endogenous SIRT-1 is a protective mechanism in cardiac diseases34,35. Moreover, the cardio-protection of RES by activating SIRT-1 was also detected in this paper (Figure S6, S7 and Table S2), which further confirmed the beneficial roles of SIRT-1 in heart diseases. Since both PARP-1 and SIRT-1 are present in the nuclear compartment, the idea that they may compete for the common NAD+ substrate arose 10 years ago36. PARP-1 also has a higher affinity for NAD+ than does SIRT-1 (Km 20–60 μmol/L vs 100–300 μmol/L, respectively)37,38. Therefore, over-activation of PARP-1 can easily limit the NAD+ availability for the activity of SIRT-1. In this study, overactivation of PARP-1 induced by Ang II in C57BL/6J mice did lead to significant consumption of NAD+ and, subsequently, the down-regulation and inactivity of SIRT-1, as expected. More importantly, these phenotypes induced by Ang II were all reversed after the inhibition of PARP-1 activity by 6014. We confirmed that the decrease of NAD+ consumption from the strong inhibition of PARP-1's activity by 6014 was one reason for reversing the activity of SIRT-1, which benefited the protection from cardiac remodeling and dysfunction.
Interestingly, we still observed the restorative effects of SIRT-1 in the case in which the activity of PARP-1 was clearly inhibited but the NAD+ content was not reversed by 6014 at the dose of 30 mg·kg−1·d−1. Notably, the interconnection between SIRT-1 and PARP-1 is more intricate39, even though NAD+ was reported as one accepted crosslink between them. Our results also confirmed the direct interaction of SIRT-1 and PARP-1 by co-IP assay and, surprisingly, detected the expression of PARylation of SIRT-1, which was greatly up-regulated by Ang II but down-regulated by 6014 at the dose of 30 mg·kg−1·d−1. Therefore, we deduced that the PARylation of SIRT-1 might contribute to the decrease of SIRT-1 activity, and the decreased PARylation of SIRT-1 by 6014 may be another new reason for activating SIRT-1 under conditions of limited NAD+ content; we aim to investigate the novel interaction of SIRT-1 and PARP-1 in our later work.
In summary, our study confirms that PARP-1 inhibition may suggest a therapeutic strategy for cardiac diseases. The novel PARP-1 inhibitor 6014 protected mice against Ang II-induced cardiac remodeling, and ameliorated cardiac function. Treatment with 6014 might be a potential therapeutic strategy for heart diseases.
Author contribution
Pei-qing LIU and Shao-rui CHEN defined the research theme and revised the manuscript critically; Guo-shuai FENG, Cui-ge ZHU, Pan-xia WANG, Yi HUANG, Min LIU, Zhuo-ming LI, Ping HE, and Lan-lan LOU performed the research; Guo-shuai FENG, Cui-ge ZHU, Zhuo-ming LI, Shao-rui CHEN, and Pei-qing LIU analyzed the data; and Guo-shuai FENG wrote the paper.
References
Cohn JN, Ferrari R, Sharpe N . Cardiac remodeling-concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. Behalf of an International Forum on Cardiac Remodeling. J Am Coll Cardiol 2000; 35: 569–82.
Weber K, Brilla CG . Pathological hypertrophy and cardiac interstitium. Fibrosis and renin-angiotensin-aldosterone system. Circulation 1991; 83: 1849–65.
Huentelman MJ, Grobe JL, Vazquez J, Stewart JM, Mecca AP, Katovich MJ, et al. Protection from angiotensin II-induced cardiac hypertrophy and fibrosis by systemic lentiviral delivery of ACE2 in rats. Exp Physiol 2005; 90: 783–90.
Lim D-S, Lutucuta S, Bachireddy P, Youker K, Evans A, Entman M, et al. Angiotensin II blockade reverses myocardial fibrosis in a transgenic mouse model of human hypertrophic cardiomyopathy. Circulation 2001; 103: 789–91.
Paradis P, Dali-Youcef N, Paradis FW, Thibault G, Nemer M . Overexpression of angiotensin II type I receptor in cardiomyocytes induces cardiac hypertrophy and remodeling. Proc Natl Acad Sci U S A 2000; 97: 931–6.
Ichihara S, Senbonmatsu T, Price E, Ichiki T, Gaffney FA, Inagami T . Angiotensin II type 2 receptor is essential for left ventricular hypertrophy and cardiac fibrosis in chronic angiotensin II-induced hypertension. Circulation 2001; 104: 346–51.
Xu S, Bai P, Little PJ, Liu P . Poly (ADP-ribose) polymerase 1 (PARP1) in atherosclerosis: from molecular mechanisms to therapeutic implications. Med Res Rev 2014; 34: 644–75.
Schreiber V, Dantzer F, Ame JC, De Murcia G . Poly (ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol 2006; 7: 517–28.
Suzuki H, Quesada P, Farina B, Leone E . In vitro poly (ADP-ribosyl) ation of seminal ribonuclease. J Biol Chem 1986; 261: 6048–55.
Balakumar P, Singh M . Possible role of poly(ADP-ribose) polymerase in pathological and physiological cardiac hypertrophy. Methods Find Exp Clin Pharmacol 2006; 28: 683–9.
Pillai JB, Gupta M, Rajamohan SB, Lang R, Raman J, Gupta MP . Poly (ADP-ribose) polymerase-1-deficient mice are protected from angiotensin II-induced cardiac hypertrophy. Am J Physiol Heart Circ Physiol 2006; 291: H1545–H53.
Huang D, Yang C, Wang Y, Liao Y, Huang K . PARP-1 suppresses adiponectin expression through poly (ADP-ribosyl) ation of PPARγ in cardiac fibroblasts. Cardiovasc Res 2009; 81: 98–107.
Pacher P, Liaudet L, Bai P, Virag L, Mabley JG, Hasko G, et al. Activation of poly(ADP-ribose) polymerase contributes to development of doxorubicin-induced heart failure. J Pharmacol Exp Ther 2002; 300: 862–7.
Pillai JB, Russell HM, Raman J, Jeevanandam V, Gupta MP . Increased expression of poly(ADP-ribose) polymerase-1 contributes to caspase-independent myocyte cell death during heart failure. Am J Physiol Heart Circ Physiol 2005; 288: H486–96.
Bartha E, Solti I, Kereskai L, Lantos J, Plozer E, Magyar K, et al. PARP inhibition delays transition of hypertensive cardiopathy to heart failure in spontaneously hypertensive rats. Cardiovasc Res 2009; 83: 501–10.
Palfi A, Toth A, Hanto K, Deres P, Szabados E, Szereday Z, et al. PARP inhibition prevents postinfarction myocardial remodeling and heart failure via the protein kinase C/glycogen synthase kinase-3β pathway. J Mol Cell Cardiol 2006; 41: 149–59.
Szabó G, Bährle S, Stumpf N, Sonnenberg K, Szabó É, Pacher P, et al. Poly (ADP-ribose) polymerase inhibition reduces reperfusion injury after heart transplantation. Circ Res 2002; 90: 100–6.
Liu M, Li Z, Chen GW, Li ZM, Wang LP, Ye JT, et al. AG-690/11026014, a novel PARP-1 inhibitor, protects cardiomyocytes from Ang II-induced hypertrophy. Mol Cell Endocrinol 2014; 392: 14–22.
Koh SH, Chang DI, Kim HT, Kim J, Kim MH, Kim KS, et al. Effect of 3-aminobenzamide, PARP inhibitor, on matrix metalloproteinase-9 level in plasma and brain of ischemic stroke model. Toxicology 2005; 214: 131–9.
Nguewa PA, Fuertes MA, Cepeda V, Alonso C, Quevedo C, Soto M, et al. Poly(ADP-ribose) polymerase-1 inhibitor 3-aminobenzamide enhances apoptosis induction by platinum complexes in cisplatin-resistant tumor cells. Med Chem 2006; 2: 47–53.
Zhou SG, Zhou SF, Huang HQ, Chen JW, Huang M, Liu PQ . Proteomic analysis of hypertrophied myocardial protein patterns in renovascularly hypertensive and spontaneously hypertensive rats. J Proteome Res 2006; 5: 2901–8.
Yu SS, Cai Y, Ye JT, Pi RB, Chen SR, Liu PQ, et al. Sirtuin 6 protects cardiomyocytes from hypertrophy in vitro via inhibition of NF-kappaB-dependent transcriptional activity. Br J Pharmacol 2013; 168: 117–28.
Scovassi AI . Mitochondrial poly(ADP-ribosylation): from old data to new perspectives. FASEB J 2004; 18: 1487–8.
Du L, Zhang X, Han YY, Burke NA, Kochanek PM, Watkins SC, et al. Intra-mitochondrial poly(ADP-ribosylation) contributes to NAD+ depletion and cell death induced by oxidative stress. J Biol Chem 2003; 278: 18426–33.
Luo X, Kraus WL . On PAR with PARP: cellular stress signaling through poly(ADP-ribose) and PARP-1. Genes Dev 2012; 26: 417–32.
Grobe JL, Mecca AP, Lingis M, Shenoy V, Bolton TA, Machado JM, et al. Prevention of angiotensin II-induced cardiac remodeling by angiotensin-(1–7). Am J Physiol 2007; 292: H736–H42.
Mukhopadhyay P, Rajesh M, Cao Z, Horvath B, Park O, Wang H, et al. Poly (ADP-ribose) polymerase-1 is a key mediator of liver inflammation and fibrosis. Hepatology (Baltimore, Md) 2014; 59: 1998–2009.
Hu B, Wu Z, Hergert P, Henke CA, Bitterman PB, Phan SH . Regulation of myofibroblast differentiation by poly(ADP-ribose) polymerase 1. Am J Pathol 2013; 182: 71–83.
Huang D, Wang Y, Yang C, Liao Y, Huang K . Angiotensin II promotes poly(ADP-ribosyl)ation of c-Jun/c-Fos in cardiac fibroblasts. J Mol Cell Cardiol 2009; 46: 25–32.
Wang Y, Wang L, Zhang F, Zhang C, Deng S, Wang R, et al. Inhibition of PARP prevents angiotensin II-induced aortic fibrosis in rats. Int J Cardiol 2013; 167: 2285–93.
Yamamoto H, Schoonjans K, Auwerx J . Sirtuin functions in health and disease. Mol Endocrinol 2007; 21: 1745–55.
Bindu S, Pillai VB, Gupta MP . Role of sirtuins in regulating pathophysiology of the heart. Trends Endocrinol Metab 2016; 27: 563–73.
Winnik S, Auwerx J, Sinclair DA, Matter CM . Protective effects of sirtuins in cardiovascular diseases: from bench to bedside. Eur Heart J 2015; 36: 3404–12.
Thandapilly SJ, Wojciechowski P, Behbahani J, Louis XL, Yu L, Juric D, et al. Resveratrol prevents the development of pathological cardiac hypertrophy and contractile dysfunction in the SHR without lowering blood pressure. Am J Hypertens 2010; 23: 192–6.
Cappetta D, Esposito G, Piegari E, Russo R, Ciuffreda LP, Rivellino A, et al. SIRT1 activation attenuates diastolic dysfunction by reducing cardiac fibrosis in a model of anthracycline cardiomyopathy. Int J Cardiol 2016; 205: 99–110.
Zhang J . Are poly(ADP-ribosyl)ation by PARP-1 and deacetylation by Sir2 linked? Bioessays 2003; 25: 808–14.
Bai P, Cantó C, Oudart H, Brunyánszki A, Cen Y, Thomas C, et al. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab 2011; 13: 461–8.
Rajamohan SB, Pillai VB, Gupta M, Sundaresan NR, Birukov KG, Samant S, et al. SIRT1 promotes cell survival under stress by deacetylation-dependent deactivation of poly (ADP-ribose) polymerase 1. Mol Cell Biol 2009; 29: 4116–29.
Xu S, Bai P, Little PJ, Liu P . Poly(ADP-ribose) polymerase 1 (PARP1) in atherosclerosis: from molecular mechanisms to therapeutic implications. Med Res Rev 2014; 34: 644–75.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (No 81673433, No 81026548 and No 81273499 to Dr Pei-qing LIU), Team item of the Natural Science Foundation of Guangdong Province (No S2011030003190 to Dr Pei-qing LIU), the Major Project of Guangdong Province (No 2015B020232009, No 2014B020210003, 2013B090700010 to Dr Pei-qing LIU), the Fund of the Guangdong Provincial Bureau of Traditional Chinese Medicine (No 20161049 to Dr Shao-rui CHEN), and the Medical Scientific Research Foundation of Guangdong Province (No A2015220 to Dr Shao-rui CHEN).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Supplementary information are available at the website of Acta Pharmacologica Sinica.
Supplementary information
Supplementary Figure S1
1H NMR of AG-690/11026014 (DOC 1552 kb)
Supplementary Figure S2
13C NMR of AG-690/11026014 (DOC 891 kb)
Supplementary Figure S3
HRMS of AG-690/11026014 (DOC 71 kb)
Supplementary Figure S4
HPLC of AG-690/11026014 (DOC 55 kb)
Supplementary Figure S5
Cell viability of H9C2 cells in the presence of 6014 and 3-AB with various concentrations (0, 6.25, 12.5, 25.0, 50.0, 100, 200 μM) for 48 hours. (DOC 27 kb)
Supplementary Figure S6
RES (100 mg·kg−1·day−1) attenuated cardiac hypertrophy and fibrosis induced by AngII (1000 ng·kg−1·min−1, 4 weeks). (DOC 4383 kb)
Supplementary Figure S7
A–B, RES (100 mg·kg−1·day−1) significantly reversed the decrease of SIRT-1 protein levels and SIRT-1 activity. (DOC 1459 kb)
Supplementary Table S1
Analysis of cardiac function of C57 BL/6 mice by echocardiography after various treatments for 4 weeks. (DOC 48 kb)
Supplementary Table S2
Analysis of cardiac function of C57 BL/6 mice by echocardiography after treating with AngII and RES for 4 weeks. (DOC 38 kb)
Rights and permissions
About this article

Cite this article
Feng, Gs., Zhu, Cg., Li, Zm. et al. Synthesis of the novel PARP-1 inhibitor AG-690/11026014 and its protective effects on angiotensin II-induced mouse cardiac remodeling. Acta Pharmacol Sin 38, 638–650 (2017). https://doi.org/10.1038/aps.2016.159
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2016.159
