
Abstract
Proprotein convertase subtilisin/kexin type 9 (PCSK9), also known as neural apoptosis regulated convertase (NARC1), is a key modulator of cholesterol metabolism. PCSK9 increases the serum concentration of low-density lipoprotein cholesterol by escorting low-density lipoprotein receptors (LDLRs) from the membrane of hepatic cells into lysosomes, where the LDLRs are degraded. Owing to the importance of PCSK9 in lipid metabolism, considerable effort has been made over the past decade in developing drugs targeting PCSK9 to lower serum lipid levels. Nevertheless, some problems and challenges remain. In this review we first describes the structure and function of PCSK9 and its gene polymorphisms. We then discuss the various designs of pharmacological targets of PCSK9, including those that block the binding of PCSK9 to hepatic LDLRs (mimetic peptides, adnectins, and monoclonal antibodies), inhibit PCSK9 expression (the clustered regularly interspaced short palindromic repeats/Cas9 platform, small molecules, antisense oligonucleotides, and small interfering RNAs), and interfere with PCSK9 secretion. Finally, this review highlights future challenges in this field, including safety concerns associated with PCSK9 monoclonal antibodies, the limited utility of PCSK9 inhibitors in the central nervous system, and the cost-effectiveness of PCSK9 inhibitors.
Similar content being viewed by others


Introduction
Cardiovascular diseases that occur in patients who have high levels of low-density lipoprotein cholesterol (LDL-C) are a leading cause of death in developed countries1,2. Increased levels of LDL-C are considered a major risk factor for coronary artery disease (CHD) and for the development of atherosclerotic plaques in arteries3,4. Cardiovascular risk is decreased by 22% when LDL-C is reduced by 24 mg/dL or 38.5 mg/dL5,6.
Loss-of-function mutations in the low-density lipoprotein receptor (LDLR) gene in patients with familial hypercholesterolemia (FH) are associated with high plasma LDL-C levels and early-onset CHD, which begins in childhood7,8. The LDLR, which is localized to the cell membrane, degrades the plasma LDL-C concentration via the receptor-mediated uptake of LDL-C into the cell. FH patients lacking LDLR mutations have apolipoprotein B (apoB) gene mutations9,10,11. Specific mutations in the apoB gene increase the LDL-C levels and induce early-onset CHD by blocking the interaction between LDLRs and apoB.
One of the greatest advances in the lipid-lowering field over the past decade was the development of a lipid-altering therapy targeting proprotein convertase subtilisin/kexin type 9 (PCSK9), which binds to LDLRs and targets them for lysosomal degradation12,13. The administration of statins to reduce LDL-C has also effectively diminished the risk of cardiovascular disease over the past 20 years14,15. However, some patients, especially those with FH, are unable to achieve normal LDL-C levels, even when they are treated with the maximum dose of statins16. This condition is due to the increase in the transcription of both PCSK9 and LDLR upon using statins, which leads to a decreased lipid restriction of statins17,18,19. Therefore, agents that target PCSK9 are promising therapeutic targets for the treatment of hypercholesterolemia and might improve the efficacy of statins in CHD.
This paper summarizes current advancements in PCSK9 inhibitors, including blocking the combination of PCSK9 with hepatic LDLRs (mimetic peptides, adnectins, and monoclonal antibodies (mAbs)), inhibiting PCSK9 expression (the clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 genome editing platform, small molecules, antisense oligonucleotides, and small interfering RNAs (siRNAs)), and interfering with PCSK9 secretion. This review also discusses the opportunities and challenges that are associated with the development of drugs that target PCSK9.
Structure, function, and gene polymorphisms of PCSK9
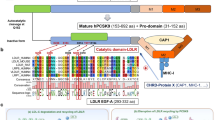
PCSK9, also known as neural apoptosis-regulated convertase 1 (NARC-1), was first described in 200320. The human PCSK9 gene is located on chromosome 1p32.3 and is 3617 bps in length. It contains 12 exons that encode 692 amino acids21,22. The PCSK9 protein contains a signal peptide, a prodomain, a catalytic domain, and a C-terminal cysteine-histidine–rich domain that is composed of 3 modules (M1, M2, and M3)23,24,25. It has been shown that PCSK9 zymogen processing and secretion are inhibited by the absence of M1 and M3. By contrast, M2 is not required for PCSK9 maturation and release, but it is important for intracellular LDLR degradation. The molecular weight of the PCSK9 precursor protein is 75 kDa. After autocatalytic cleavage in the endoplasmic reticulum (ER), the prodomain is separated from the 62 kDa mature PCSK9 protein. The separated prodomain remains non-covalently bound to the mature PCSK9 protein, thus forming a prosegment-PCSK9 complex that forces the PCSK9 catalytic domain into an inactive conformation26. The cleaved complex is then transported from the ER to the Golgi apparatus and then released27. The secreted PCSK9 then interacts via its catalytic domain28 and prodomain29 with the epidermal growth factor-like repeat homology domain-A (EGF-A) and β-propeller domain, respectively, of the LDLR. The binding of PCSK9 to the LDLR is blocked by mutations in the EGF-A of the LDLR, which is a major binding site for PCSK912. Recently, it has been reported that the interaction between the C-terminal domain (CTD) of PCSK9 and the ligand-binding domain (LBD) of the LDLR may be enhanced in an acidic milieu30. In addition, it has been shown that the PCSK9 CTD is necessary for the localization of PCSK9 to the trans-Golgi network (TGN) and that it increases the endocytosis of the PCSK9/LDLR complex31. To date, the mechanism by which the PCSK9/LDLR complex is targeted for degradation is unclear, although the available evidence suggests that at least two mechanisms are possible. First, the affinity between the PCSK9 catalytic domain and LDLR EGF-A is increased under acidic conditions; this could change the conformation of the LDLR, thereby promoting its degradation in lysosomes after formation of the PCSK9/LDLR complex31. Second, there is evidence to indicate that transmembrane protein X targets the PCSK9/LDLR complex to lysosomes for degradation by forming a bridge between the PCSK9 CTD and cytosolic adaptor proteins32.
Recently, new strategies for inhibiting PCSK9 for the treatment of hypercholesterolemia have been developed. Initially, mAbs that change the conformation of the catalytic domain of PCSK9 were discovered32. Other studies have shown that annexin A233 and a mAb34 can inhibit PCSK9-induced LDLR degradation by binding specifically to the CTD of PCSK9.
The synthesis, secretion, and expression of PCSK9 occur primarily in the liver; PCSK9 is expressed at low levels in the gastrointestinal tract, kidneys, and central nervous system (CNS)20,21. In hepatic cells, secreted PCSK9 can bind to LDLRs in the hepatocellular membrane through the EGF-A of the LDLR. The PCSK9/LDLR complex enters the endosomal system and is then degraded in the lysosomes before LDLR transport to the cell surface35,36. Therefore, increased levels of PCSK9 can inhibit LDLR recycling to the cell surface35,36,37, which can in turn increase the LDL-C levels in the bloodstream. Recent studies have shown that PCSK9 might modulate LDLR transport by interacting with amyloid precursor-like protein 2, a protein that transports transmembrane proteins to the lysosomes38,39,40 (Figure 1). In the small intestine, PCSK9 is closely associated with apoB, which is important in triglyceride and postprandial lipoprotein production41. In one study, the secretion of apoB48 and apoB100 was increased by 40% and 55%, respectively, in human enterocytes (Caco-2 cells) treated with human PCSK941. Transintestinal cholesterol excretion is also modulated by PCSK942. In addition to the LDLR, PCSK9 can mediate the degradation of other lipid receptors in the LDLR family, such as the very low-density lipoprotein receptor43, the apolipoprotein E receptor 243, and LDL receptor-related protein 1. PCSK9 also appears to play an important role in the pathogenesis of diseases such as diabetes44, cancer45, and Alzheimer's disease46,47.

Steps in the LDLR metabolic pathway. After LDL binds to the LDLR at the cell membrane, the LDLR-LDL complex undergoes endocytosis. Within the endosome, LDL dissociates from the LDLR. The LDL then enters the lysosome where it is degraded, and the LDLR is recycled to the cell surface. The LDLR metabolic pathway under PCSK9 regulation. Secreted PCSK9 binds to the LDLR at the cell surface, and the PCSK9-LDLR complex is endocytosed, thereby promoting its lysosomal degradation. PCSK9-targeted therapies include mAbs, adnectins, and mimetic peptides (which block the interaction between PCSK9 and the LDLR), CRISPR/Cas9 genome editing technology, antisense oligonucleotides (ASOs), siRNAs and small molecules (which inhibit PCSK9 expression), and drugs that interfere with PCSK9 secretion from the ER.
A mutation in the PCSK9 gene was first detected in 2003 in French families48 and PCSK9 was the third gene to be linked to autosomal dominant FH, after apoB and LDLR. In autosomal dominant FH, a gain-of-function mutation in the PCSK9 gene results in elevated levels of LDL-C48,49,50,51,52. In addition, overexpressing PCSK9 in the hepatic cells of mice elevated the levels of total plasma and non-high-density lipoprotein cholesterol53 and decreased the levels of LDLR in hepatic cells54, which indicated that the increased risk of cardiovascular disease is due to PCSK9 gain-of-function mutations in humans. In 2005, loss-of-function mutations in PCSK9 were found in people who had lower LDL-C levels and a significantly reduced incidence of cardiovascular events. Two nonsense mutations (Y142X and C679X) of the coding region of PCSK9, which were associated with reduced LDL-C in 128 participants (50% African American), were reported in a study. The incidences of these two mutations were more common in African Americans compared with European Americans and were correlated with a 40% decrease in the plasma concentration of LDL cholesterol55. Moreover, PCSK9 loss-of-function patients were shown to have a heightened response to statin therapy. Thus, PCSK9 inhibitors are required to enhance the lowering action of LDL-C of statins56.
PCSK9 as an emerging target for LDL-C-lowering therapy
Researchers have devoted much time and knowledge to examining PCSK9 as a significant therapeutic target. Such research primarily includes blocking the combination of PCSK9 and LDLR via mAbs, adnectins, and mimetic peptides, inhibiting PCSK9 expression with CRISPR/Cas9 genome-editing technology, antisense oligonucleotides (ASOs) and siRNA, and interfering with PCSK9 secretion from the ER (Figure 1, Table 1).
Blocking the PCSK9-LDLR combination
Mimetic peptides
Mimetic peptides that inhibit the interaction between PCSK9 and the LDLR include several short amino acid sequences that mimic the EGF-A35,57,58, catalytic domain, prodomain, or C-terminal domain of PCSK925,59,60. These therapeutic small peptides have high specificity, are easy to produce and modify, and are cheaper to produce than antibodies, but their routes of administration are limited.
Adnectins
Adnectins are a new type of therapeutic protein based on the 10th fibronectin type III domain. They bind to their target proteins with high specificity and affinity via transforming β-sheet loops, which provide structural stability. BMS-962476 (Bristol-Myers Squibb/Adnexus) is thought to prevent PSCK9-mediated LDLR degradation61.
Antibodies targeting PCSK9
Currently, mAb administration is an effective therapeutic way to suppress the interaction between PCSK9 and LDLR because of its high titer, high specificity, and long half-life. Primary mAbs that have been developed against PCSK9 include alirocumab (REGN727/SAR236553), evolocumab (AMG 145), and bococizumab (RN316). Antibodies that target a single domain of PCSK9 are powerful inhibitors that mediate the degradation of the LDLR62.
Alirocumab (REGN727/SAR236553) developed by Sanofi/Regeneron
Alirocumab (REGN727/SAR236553), which is marketed by Sanofi/Regeneron under the brand name Praluent, is a human mAb that inhibits PCSK9 63. Its weight is approximately 146 kDa. Within 4–8 h, alirocumab achieves maximum suppression and is dose-dependent. Within 3–7 d, the maximum serum concentration occurs via the subcutaneous injection of alirocumab, which is 75 mg or 150 mg every 2 weeks, and its bioavailability increases by approximately 85%. The half-life of alirocumab in patients who are intolerant to statins and have heterozygous FH (HeFH) is 17–20 d. In Phase II trials, LDL-C was reduced by 40%–70% through the combined treatment of alirocumab and statins64,65,66. The LDL-C levels were reduced to less than 100 mg/dL in all patients treated with SAR236553 compared with 52% of patients treated with 80 mg of atorvastatin plus placebo. Similarly, the LDL-C levels were reduced to less than 70 mg/dL in 90% of patients treated with SAR236553 compared with 17% of patients treated with 80 mg of atorvastatin plus placebo65. Three Phase III Odyssey FH studies found that LDL-C levels were reduced by an additional 49% in patients treated with alirocumab compared with untreated patients67. Across all trials, LDL-C levels of <70 mg/dL were achieved in the majority of patients treated with alirocumab68.
Evolocumab (AMG 145) developed by Amgen
Evolocumab (AMG 145), which is marketed by Amgen under the brand name Repatha, is a human monoclonal antibody that inhibits PCSK969. Repatha has a molecular weight of 144 kDa. Evolocumab is administered by subcutaneous injection either biweekly or monthly, at a dose of 140 mg or 420 mg, respectively, and causes a dose-dependent reduction in LDL-C. Evolocumab has a bioavailability of approximately 72%, and a half-life of 11–17 d. After 12 weeks of dosing, a steady state was reached. In Phase I and II trials, a subcutaneous injection of evolocumab reduced the LDL-C levels in HeFH and non-FH patients; the apoB and lipoprotein (a) levels were also reduced by 30%–59% and 18%–36%, respectively, in a dose-dependent manner70. In a meta-analysis of Phase II and III trials, evolocumab and alirocumab reduced LDL-C and lipoprotein (a) levels by 47% and 26%, respectively71. The Phase III Descartes study, which determined the efficacy and safety of evolocumab in hyperlipidemic patients, showed that evolocumab reduced LDL-C by 48.5%–61.6%72. Evolocumab reduced LDL-C by <70 mg/dL in 82% of subjects73. In Phase II and III trials, adverse reactions (including nasopharyngitis, headache, upper respiratory tract infection, diarrhea, rash, myalgia, back pain, and urticaria), were reported at similar levels among patients receiving evolocumab treatment or placebo. Currently, evolocumab is prescribed primarily to patients who have FH and are intolerant to statin therapy.
Bococizumab (RN316) developed by Pfizer
Bococizumab (RN316) is another humanized mAb that directly inhibits PCSK9. A Phase II, randomized, placebo-controlled study by Gumbiner et al determined the efficacy and safety of bococizumab in hypercholesterolemic patients receiving high-dose statin therapy74. After 12 weeks, bococizumab administration decreased LDL-C by 56%, compared with 4% in the placebo group. In several patients receiving bococizumab, LDL-C was reduced to levels below 25 mg/dL, leading to an interruption in treatment at week 4. Bococizumab is more potent than other LDL-C-lowering mAbs. In a randomized, placebo-controlled trial, 150 mg of bococizumab biweekly reduced the LDL-C levels by 53%75. Adverse events were reported at similar levels in patients receiving bococizumab or placebo. The SPIRE program is currently conducting five Phase III trials with bococizumab (SPIREHF, SPIRE-LDL, SPIRE-HR, SPIRE-1, and SPIRE-2).
Inhibition of PCSK9 expression
CRISPR/Cas9 platform
CRISPR-Cas9, a novel genome editing technology, is based on the CRISPR adaptive immune system of bacteria and comprises a guided RNA linked to an endonuclease (ie, Cas9). Recently, the CRISPR-Cas9 platform was found in a program to specifically target and cleave DNA using a single-stranded RNA molecule and has thus received considerable attention11,76. Many bacteria use the CRISPR system to protect themselves against the invasion of foreign nucleic acids, including viruses and plasmids. Activation of the CRISPR-Cas9 mechanism induces double-stranded breaks in the DNA of host cells, followed by error-prone recombination and non-homologous end-joining. This process produces frameshift mutations and allele knockout following the infection of CRISPR-Cas9-containing host cells with viruses or plasmids77. The mutagenesis rate of PCSK9 in mouse liver was found to be >50% on days three through four, and off-target mutagenesis was minimized by treatment with adenovirus to express Cas9 and a CRISPR guide RNA targeting PCSK9. This approach reduced the PCSK9 levels, increased the hepatic LDL receptor levels, and reduced the plasma cholesterol levels by 35%–40%77. In a study using Fah−/−Rag2−/−Il2rg−/− (FRG KO) mice, which have chimeric, humanized livers, treatment with CRISPR-Cas9 targeting the human PCSK9 gene induced high levels of on-target mutagenesis of PCSK9, resulting in a 52% decrease in the level of human PCSK9 protein in the blood; off-target mutagenesis was not detected78. A compensatory mechanism within mouse hepatocytes, which increases the post-treatment blood levels of mouse PCSK9 protein more than twofold, has been found in transplanted FRG KO mice. Although the CRISPR-Cas9 platform that targets PCSK9, which can cause a permanent PCSK9 alteration, is a promising tool editing gene for use in humans, many questions remain unanswered. Considering the chimeric liver-humanized FRG KO mouse with default immunity and the loss of immune consequences of viral vectors targeting human PCSK9 in the mice liver, a future strategy may aim for the double humanization of mice-related FRG KO to both the liver and hematopoietic systems79.
Small-molecule inhibitors
Small molecules have also received considerable attention. Unfortunately, efforts to develop small-molecule inhibitors of PCSK9, which are desirable because of their low cost, have been unsuccessful. Pep2-8, a peptide-based inhibitor of PCSK9 that mimics the secondary structural elements of the EGF-A domain of the LDLR, binds to PCSK9 and competitively inhibits the binding of PCSK9 to the LDLR. The major challenge associated with the development of PCSK9 small-molecule inhibitors is the lack of small-molecule-binding sites on PCSK9 due to the relatively flat surface of PCSK9; by contrast, antibody-binding sites are readily accessible. Regardless, other small molecules hold promise, such as berberine and oleanolic acid, which directly inhibit PCSK9 expression80.
Berberine (BBR)
BBR is a type of protoberberine that is also known as an isoquinoline plant alkaloid. BBR elicits several pharmacological effects through diverse mechanisms, including antagonizing microbes and tumors, immunomodulation, and lowering both glucose and cholesterol81,82. Accumulating evidence indicates that there is a close relationship between BBR and the PCSK9-LDLR pathway, an important mediatory of cholesterol depletion83,84. In liver tissue, transcription factors, including members of the sterol regulatory element-binding protein (SREBP) family (which bind to the SRE motif in the proximal promoter) and hepatocyte nuclear factor 1α (HNF1α; an essential trans-activator containing homeodomain)85, co-regulate the transcription of PCSK986. The original study found that the SREBP pathway did not involve a reduction of the PCSK9 mRNA levels induced by BBR; specifically, the levels of SREBP-2 mRNA increased by 74%, and the question of whether BBR could decrease the activity of PCSK9 promoter was addressed84. In subsequent studies, BBR was found to simultaneously decrease the levels of HNF1α (a transcription factor that specifically recognizes the site on the PCSK9 promoter between Sp1 and SRE) and SREBP2. Thus, the interaction of two transcription factor with their binding sequence of PCSK9 promoter is reduced, and PCSK9 transcription is repressed87. Recently, in vivo studies using hyperlipidemic mice suggested that the serum PCSK9 concentrations decreased by 50%, that the mRNA level of PCSK9 in all liver samples decreased by 46%, and that the LDLR protein levels increased by 67% in the BBR compared with the control, whereas the mRNA levels of LDLR and other SREBP2 target genes were unchanged86. BBR treatment reduced HNF1α protein levels by 42% compared with control, whereas HNF1α mRNA levels were unaffected by BBR treatment. These results were consistent with those obtained for a hyperlipidemic hamster. By blocking the ubiquitin proteasome system (UPS) and the autophagy–lysosomal pathway, BBR inhibited HNF1α-mediated PCSK9 transcription via UPS to decrease the hepatic HNF1α protein levels without affecting the mRNA levels86. Several studies have also reported that BBR has neuroprotective effects (such as suppressed neuronal apoptosis88 and neuroinflammation89) and both anti-apoptotic and anti-inflammatory effects90,91. Additional studies are needed to determine whether BBR exerts its effects through PCSK9.
Oleanolic acid (OA)
OA is a pentacyclic triterpene that is widely distributed in plants and medicinal herbs as a free acid or as a saponin aglycone92,93. OA has numerous beneficial properties, including anti-cancer, hepatoprotective, hypolipidemic94,95, antioxidative, anti-inflammatory96, endothelial protective97, and anti-atherosclerotic effects.
In OA-treated db/db mice, serum triglycerides, total cholesterol (TC), LDL-cholesterol, free fatty acids, and the quantity of lipid droplets in hepatic cells were markedly reduced compared with untreated mice98. Furthermore, our previous studies have shown that OA decreases the levels of PCSK9 protein and mRNA in HepG2 cells, in a time- and dose-dependent manner99. However, the underlying mechanism is unknown, and the OA efficiency is limited because of its low bioavailability and insolubility in water.
Antisense oligonucleotides (ASOs)
ASOs, which interfere with mRNA activation, consist of short, single-stranded nucleotide sequences. The successful delivery of ASOs to the hepatic nucleus has been reported100. By binding to their target mRNA, ASOs prevent protein translation and thereby reduce protein levels. In one study, the administration of an ASO (ISIS 394814) to hyperlipidemic mice for 6 weeks demonstrated that the levels of PCSK9 mRNA and LDL-C were reduced by 92% and 32%, respectively, that TC was reduced by 52%, and that the LDLR protein levels were increased twofold101. In addition, two locked antisense oligonucleotides (SPC5001 and SPC4061) targeting PCSK9 decreased the levels of plasma PCSK9 and LDL-C by 85% and 50%, respectively. A Phase I clinical trial on BMS-844421 was terminated because of safety concerns67. Both ends of ASO (SPC5001) DNA are locked with RNA nucleotides, which are composed of one monomer and are stable102. Even if ASO has high affinity and specificity, the high production cost and required routes for intravenous or subcutaneous administration limit its use in individuals with hyperlipidemia.
siRNA
The intravenous administration of single-chain siRNAs in lipid nanoparticles is a new therapeutic approach to inhibiting PCSK9 activity103. Studies in mice and rats have reported that siRNA-induced PCSK9 silencing decreased the PCSK9 mRNA levels by 50%–70% and the TC concentrations by 60%. Another study in non-human primates found that siRNA-mediated knockdown of PCSK9 was rapid, sustained, and reversible and that it resulted, on average, in a 56% reduction in the LDL-C levels. A Phase I clinical trial by Alnylam Pharmaceuticals (ALN-PCS) demonstrated that administration of their siRNA (ALN-PCSsc) resulted in a 70% reduction in plasma PCSK9 and a 40% reduction in LDL-C relative to baseline104. Another Phase I clinical trial of subcutaneously administered ALN-PCSsc has also been completed59. A Phase II trial of ALN-PCSsc is currently in progress58.
Interfering with PCSK9 secretion
Two specific mediators, sortilin105 and Sec24a106, are known to be involved in PCSK9 secretion. Sortilin is important in lipoprotein metabolism as a transmembrane type I transport receptor, and it is not directly regulated by PCSK9. Conversely, sortilin, which co-localizes with PCSK9 in the trans-Golgi network, facilitates PCSK9 secretion from primary hepatocytes in the late secretory pathway. Sortilin is encoded by the gene SORT1 and is a high-affinity sorting receptor for PCSK9. Sortilin thus represents a good target for the treatment of hypercholesterolemia. Plasma PCSK9 is reduced in sortilin-deficient mice but is elevated following sortilin overexpression in the liver. Moreover, a positive correlation exists between the levels of circulating PCSK9 and sortilin levels in healthy humans. One study found that the absence of Sec24a (also known as coat protein complex II adaptor protein) inhibited the early transport of PCSK9 from the ER to the cis-Golgi, leading to an increase in LDLR levels and a decrease in LDL-C levels. Moreover, SRT3025, a sirtuin 1 deacetylase activator, inhibited the development of atherosclerosis in ApoE-/- mice by decreasing PCSK9 secretion and increasing LDLR levels107.
The Q152H loss-of-function mutation in PCSK9 specifically blocked PCSK9 secretion by decreasing its intramolecular cleavage, leading to the accumulation of unprocessed proPCSK9 within the cell108.
Challenges and future perspectives
Despite the proven efficacy of PCSK9 mAbs, many challenges must be addressed in future clinical research. Special attention should be given to the side effects associated with PCSK9 mAbs. Cognitive decline may result from the deposition of soluble amyloid-β (Aβ) on the walls of cerebral capillaries and arteries, a process that is potentiated by the formation of immune complexes such as those formed between mAbs and PCSK9109. Immune complexes can also elicit complement activation through the classic C1q-mediated antibody-dependent pathway, leading to abnormal glutamate release and neurotoxicity 110.
Side effects of PCSK9 mAbs
Neurocognitive adverse effects
The incidence of neurocognitive adverse events in alirocumab-treated subjects was 0.8%, compared with 0.7% in the placebo-treated group. Alirocumab treatment resulted in memory impairment and confusion in 0.2% of patients, whereas the incidence of these side effects in the placebo groups did not reach 0.1%63. In other trials, treatment with evolocumab produced abnormally low LDL-C levels (<50 mg/dL) in <1% of patients; these trials reported side effects such as amnesia and mental impairment111.
Other adverse events in clinical trials of PCSK9 mAbs
In clinical trials of alirocumab, the incidences of other adverse events in the alirocumab group compared with the placebo group, respectively, were as follows: injection site reactions (7.2% vs 5.1%), nasopharyngitis (11.3% vs 11.1%), influenza (5.7% vs 4.6%), urinary tract infection (4.8% vs 4.6%), cough (2.5% vs 2.3%), myalgia (4.2% vs 3.4%), sinusitis (3.0% vs 2.7%), musculoskeletal pain (2.1% vs 1.6%), bronchitis (4.3% vs 3.8%), diarrhea (4.7% vs4.4%), and upper respiratory tract infection (3.1% vs 2.4%)63. In clinical trials of evolocumab, the incidences of adverse events in the evolocumab group compared with the placebo group, respectively, were as follows: nasopharyngitis (10.5% vs 9.6%), upper respiratory tract infection (9.3% vs 6.3%), influenza (7.5% vs 6.3%), back pain (6.2% vs 5.6%), injection site reactions (5.7% vs 5.0%), cough (4.5% vs3.6%), urinary tract infection (4.5% vs 3.6%), sinusitis (4.2% vs3.0%), headache (4.0% vs 3.6%), myalgia (4.0% vs 3.0%), dizziness (3.7% vs 2.6%), musculoskeletal pain (3.3% vs 3.0%), hypertension (3.2% vs 2.3%), diarrhea (3.0% vs 2.6%), and gastroenteritis (3.0% vs 2.0%). Overall, adverse events were reported more frequently in patients receiving alirocumab or evolocumab compared with placebo112 (Table 2).
Side effect profiles of nearly all antibodies
Hypersensitivity reactions
Allergic reactions, including urticaria, pruritus, and rash, as well as serious adverse events such as hypersensitivity vasculitis have been reported with alirocumab treatment. Allergic reactions were reported more frequently in patients treated with alirocumab compared with those treated with placebo (8.6% vs 7.8%, respectively). The percentage of patients who discontinued therapy because of allergic reactions was higher than that in the placebo group (0.6% vs 0.2%, respectively)35.
The incidence of allergic reactions was 5.1% in evolocumab-treated patients and 4.7% in placebo-treated patients. In general, hypersensitivity reactions (in the evolocumab vs placebo groups) involve rash (1.0% vs0.5%), erythema (0.4% vs0.2%), eczema (0.4% vs 0.2%), and urticaria (0.4% vs 0.1%)57.
Immunogenicity
Alirocumab and other therapeutic proteins carry the potential for immunogenicity. In 10 placebo-controlled studies, patients obtaining alirocumab who developed anti-drug antibodies (ADAs) were detected; and the alirocumab rate was 4.8%, whereas the control rate was 0.6%35. Subjects with ADAs had a higher incidence of injection site reactions (10.2%) compared with subjects lacking anti-drug antibodies (5.9%). Moreover, 1.2% of patients developed neutralizing antibodies (NAbs), all of which occurred in the alirocumab group. Most of these patients had only one positive neutralizing sample. Only 0.3% of patients had 2 or more NAb-positive samples. The long-term consequences of alirocumab immunogenicity require further investigation35. Additionally, 0.1% of subjects receiving evolocumab developed ADAs at least once over a series of trials. Neutralizing antibodies continue to be found in clinical trials, but their long-term effects remain unknown111.
MAbs that are used to treat cardiovascular disease, such as abciximab and inclacumab, are associated with off-target effects, non-specific adverse events, on-target adverse events, and the production of ADAs113,114,115. In our previous research, we demonstrated that PCSK9 can influence neuronal apoptosis in two ways: it can maintain apoptosis at low levels and can limit increases in apoptosis. However, mAbs against PCSK9 are of limited use in the CNS to treat nervous system diseases because of their large molecular weight (>400–600 kDa)116 and the neurotoxicity of the mAb-PCSK9 immune complex. The high cost of PCSK9 mAbs is also prohibitive. One elegant study found that adding a PCSK9 inhibitor to existing statin therapy in all eligible patients increased the annual prescription drug expenditure in the United States (US) by approximately $125 billion over ezetimibe use alone (a 38% increase from $329 billion in prescription drug expenditures) and increased US health care spending by approximately $120 billion (a 4% increase from the $2.8 trillion dollars in total US health care expenditures) in 2015117.
Future perspectives
Clinical trials have demonstrated that drugs targeting PCSK9 effectively reduce LDL-C levels. PCSK9 mAbs, including alirocumab and evolocumab, were approved for the market in 2015, and a Phase III trial of bococizumab was completed recently68. The development of other therapies, such as gene-silencing agents, small molecules (BBR and OA), mimetic peptides, adnectins, and inhibitors of PCSK9 secretion, are described in this review. A novel genome editing technology, CRISPR-Cas9, induced a high rate of PCSK9 mutagenesis of approximately 50%; this approach significantly decreased plasma PCSK9 protein levels, with a low incidence of off-target mutations118. The challenges associated with the development of drugs targeting PCSK9, including long-term safety concerns, effects on the CNS, and the cost-effectiveness of PCSK9 mAbs, are discussed at the end of this review. Despite the inhibitory effect of PCSK9 on neuronal apoptosis, the utility of PCSK9 mAbs in the CNS is limited because of their high molecular weight and the neurotoxicity of the PCSK9-mAb immune complex. Moreover, the cost of PCSK9 mAbs is prohibitive. In summary, PCSK9 inhibitors appear to have clinical value, but more work is needed to understand PCSK9 protein structure, to identify novel sites of drug action, to understand the mechanisms by which drugs target PCSK9, and to develop novel small molecule compounds that act as PCSK9 antagonists.
References
Agarwal SK, Avery CL, Ballantyne CM, Catellier D, Nambi V, Saunders J, et al. Sources of variability in measurements of cardiac troponin T in a community-based sample: the atherosclerosis risk in communities study. Clin Chem 2011; 57: 891–7.
Boekholdt SM, Arsenault BJ, Mora S, Pedersen TR, Larosa JC, Nestel PJ, et al. Association of LDL cholesterol, non-HDL cholesterol, and apolipoprotein B levels with risk of cardiovascular events among patients treated with statins: a meta-analysis. JAMA 2012; 307: 1302–9.
Jansen H, Samani NJ, Schunkert H . Mendelian randomization studies in coronary artery disease. Eur Heart J 2014; 35: 1917–24.
European Association for Cardiovascular Prevention & Rehabilitation1, Reiner Z, Catapano AL, De Backer G, Graham I, Taskinen MR, et al. ESC/EAS guidelines for the management of dyslipidaemias: the task force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and the European Atherosclerosis Society (EAS). Eur Heart J 2011; 32: 1769–818.
Cholesterol Treatment Trialists C, Baigent C, Blackwell L, Emberson J, Holland LE, Reith C, et al. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170 000 participants in 26 randomised trials. Lancet 2010; 376: 1670–81.
Sniderman A, Thanassoulis G, Couture P, Williams K, Alam A, Furberg CD . Is lower and lower better and better? A re-evaluation of the evidence from the cholesterol treatment trialists' collaboration meta-analysis for low-density lipoprotein lowering. J Clin Lipidol 2012; 6: 303–9.
Lehrman MA, Schneider WJ, Sudhof TC, Brown MS, Goldstein JL, Russell DW . Mutation in LDL receptor: Alu-Alu recombination deletes exons encoding transmembrane and cytoplasmic domains. Science 1985; 227: 140–6.
Ahmad Z, Li X, Wosik J, Mani P, Petr J, McLeod G, et al. Premature coronary heart disease and autosomal dominant hypercholesterolemia: Increased risk in women with LDLR mutations. J Clin Lipidol 2016; 10: 101–8 e1–3.
Musunuru K, Kathiresan S . Surprises from genetic analyses of lipid risk factors for atherosclerosis. Circ Res 2016; 118: 579–85.
Soria LF, Mccarthy BJ . Association between a specific apolipoprotein B mutation and familial defective apolipoprotein B-100. Proc Natl Acad Sci U S A 1989; 86: 587–91.
Abifadel M, Varret M, Rabès JP, Allard D, Ouguerram K, Devillers M, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet 2003; 34: 154–6.
Zhang DW, Lagace TA, Garuti R, Zhao Z, McDonald M, Horton JD, et al. Binding of proprotein convertase subtilisin/kexin type 9 to epidermal growth factor-like repeat A of low density lipoprotein receptor decreases receptor recycling and increases degradation. J Biol Chem 2007; 282: 18602–12.
Maxwell KN, Fisher EA, Breslow JL . Overexpression of PCSK9 accelerates the degradation of the LDLR in a post-endoplasmic reticulum compartment. Proc Natl Acad Sci U S A 2005; 102: 2069–74.
Baigent C, Keech A, Kearney PM, Blackwell L, Buck G, Pollicino C, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90 056 participants in 14 randomised trials of statins. Lancet 2005; 366: 1267–78.
Genser B, März W . Low density lipoprotein cholesterol, statins and cardiovascular events: a meta-analysis. Clin Res Cardiol 2006; 95: 393–404.
Avis HJ, Hutten BA, Gagné C, Langslet G, Mccrindle BW, Wiegman A, et al. Efficacy and safety of rosuvastatin therapy for children with familial hypercholesterolemia. J Am Coll Cardiol 2010; 55: 1121–6.
Careskey HE, Davis RA, Alborn WE, Troutt JS, Cao G, Konrad RJ . Atorvastatin increases human serum levels of proprotein convertase subtilisin/kexin type 9. J Lipid Res 2008; 49: 394–8.
Welder G, Zineh I, Pacanowski MA, Troutt JS, Cao G, Konrad RJ . High-dose atorvastatin causes a rapid sustained increase in human serum PCSK9 and disrupts its correlation with LDL cholesterol. J Lipid Res 2010; 51: 2714–21.
Frederick R, Vanessa P, Andrew I, Gillian P . Elevated PCSK9 levels in untreated patients with heterozygous or homozygous familial hypercholesterolemia and the response to high-dose statin therapy. J Am Heart Assoc 2013; 2: e000028–e28.
Naureckiene S, Ma L, Sreekumar K, Purandare U, Lo CF, Huang Y, et al. Functional characterization of Narc 1, a novel proteinase related to proteinase K. Arch Biochem Biophys 2003; 420: 55–67.
Seidah NG, Benjannet S, Wickham L, Marcinkiewicz J, Jasmin SB, Stifani S, et al. The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): Liver regeneration and neuronal differentiation. Proc Natl Acad Sci U S A 2003; 100: 928–33.
Seidah NG, Prat A . Precursor convertases in the secretory pathway, cytosol and extracellular milieu. Essays Biochem 2002; 38: 79–94.
Benjannet S, Rhainds D, Essalmani R, Mayne J, Wickham L, Jin W, et al. NARC-1/PCSK9 and its natural mutants: zymogen cleavage and effects on the low density lipoprotein (LDL) receptor and LDL cholesterol. J Biol Chem 2004; 279: 48865–75.
Cunningham D, Danley DE, Geoghegan KF, Griffor MC, Hawkins JL, Subashi TA, et al. Structural and biophysical studies of PCSK9 and its mutants linked to familial hypercholesterolemia. Nat Struct Mol Biol 2007; 14: 413–19.
Du F, Hui Y, Zhang M, Linton MF, Fazio S, Fan D . Novel domain interaction regulates secretion of proprotein convertase subtilisin/kexin type 9 (PCSK9) protein. J Biol Chem 2011; 286: 43054–61.
Seidah NG, Awan Z, Chrétien M, Mbikay M . PCSK9: a key modulator of cardiovascular health. Circ Res 2014; 114: 1022–36.
Seidah NG, Benjannet S, Wickham L, Marcinkiewicz J, Jasmin SB, Stifani S, et al. The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): liver regeneration and neuronal differentiation. Proc Natl Acad Sci U S A 2003; 100: 928–33.
Zhang DW, Garuti R, Tang WJ, Cohen JC, Hobbs HH . Structural requirements for PCSK9-mediated degradation of the low-density lipoprotein receptor. Proc Natl Acad Sci U S A 2008; 105: 13045–50.
Li J, Tumanut C, Gavigan JA, Huang WJ, Hampton EN, Tumanut R, et al. Secreted PCSK9 promotes LDL receptor degradation independently of proteolytic activity. Biochem J 2007; 406: 203–7.
Surdo PL, Bottomley MJ, Calzetta A, Settembre EC, Cirillo A, Pandit S, et al. Mechanistic implications for LDL receptor degradation from the PCSK9/LDLR structure at neutral pH. EMBO Rep 2011; 12: 1300–5.
Poirier S, Hamouda HA, Villeneuve L, Demers A, Mayer G . Trafficking dynamics of PCSK9-Induced LDLR degradation: focus on human PCSK9 mutations and C-terminal domain. Plos One 2016; 11: e0157230.
Seidah NG, Awan Z, Chretien M, Mbikay M . PCSK9: a key modulator of cardiovascular health. Circ Res 2014; 114: 1022–36.
Mayer G, Poirier S, Seidah NG . Annexin A2 is a C-terminal PCSK9-binding protein that regulates endogenous low density lipoprotein receptor levels. J Biol Chem 2008; 283: 31791–801.
Chan JC, Piper DE, Cao Q, Liu D, King C, Wang W, et al. A proprotein convertase subtilisin/kexin type 9 neutralizing antibody reduces serum cholesterol in mice and nonhuman primates. Proc Natl Acad Sci U S A 2009; 106: 9820–5.
Zhang DW, Lagace TA, Garuti R, Zhao Z, Mcdonald M, Horton JD, et al. Binding of proprotein convertase subtilisin/kexin type 9 to epidermal growth factor-like repeat A of low density lipoprotein receptor decreases receptor recycling and increases degradation. J Biol Chem 2008; 282: 18602–12.
Poirier S, Mayer G, Benjannet S, Bergeron E, Marcinkiewicz J, Nassoury N, et al. The proprotein convertase PCSK9 induces the degradation of low density lipoprotein receptor (LDLR) and its closest family members VLDLR and ApoER2. J Biol Chem 2008; 283: 2363–72.
Yan H, Ma YL, Gui YZ, Wang SM, Wang XB, Gao F, et al. MG132, a proteasome inhibitor, enhances LDL uptake in HepG2 cells in vitro by regulating LDLR and PCSK9 expression. Acta Pharmacol Sin 2014; 35: 994–1004.
Devay RM, Shelton DL, Liang H . Characterization of proprotein convertase subtilisin/kexin type 9 (PCSK9) trafficking reveals a novel lysosomal targeting mechanism via amyloid precursor-like protein 2 (APLP2). J Biol Chem 2013; 288: 10805–18.
Devay RM, Yamamoto L, Shelton DL, Liang H . Common proprotein convertase subtilisin/Kexin type 9 (PCSK9) epitopes mediate multiple routes for internalization and function. PLoS One. 2015 Apr 23;10: e0125127. doi:10.1371/journal.pone.0125127. 2015; 10: 1551–52.
Dattoli G, Torre A, Mezi L, Gallardo JC, Caloi R . Amyloid precursor-like protein 2 increases the endocytosis, instability, and turnover of the H2-K(d) MHC class I molecule. J Immunol 2008; 181: 1978–87.
Rashid S, Tavori H, Brown PE, Linton MF, He J, Giunzioni I, et al. Proprotein convertase subtilisin kexin type 9 promotes intestinal overproduction of triglyceride-rich apolipoprotein B lipoproteins through both low-density lipoprotein receptor-dependent and -independent mechanisms. Circulation 2014; 130: 431–41.
Ae VDV, Vrins CL . Van dOK, Kunne C, Oude Elferink RP, Kuipers F, et al. Direct intestinal cholesterol secretion contributes significantly to total fecal neutral sterol excretion in mice. Gastroenterology 2007; 133: 967–75.
Poirier S, Mayer G, Benjannet S, Bergeron E, Marcinkiewicz J, Nassoury N, et al. The proprotein convertase PCSK9 induces the degradation of low density lipoprotein receptor (LDLR) and its closest family members VLDLR and ApoER2. J Biol Chem 2008; 283: 2363–72.
Miao J, Manthena PV, Haas ME, Ling AV, Shin DJ, Graham MJ, et al. Role of insulin in the regulation of proprotein convertase subtilisin/kexin type 9. Arteriosclerosis Thromb Vasc Biol 2015; 35: 1589–96.
Huang J, Li L, Lian J, Schauer S, Vesely PW, Kratky D, et al. Tumor-induced hyperlipidemia contributes to tumor growth. Cell Rep 2016; 15: 336–48.
Jonas MC, Costantini C, Puglielli L . PCSK9 is required for the disposal of non-acetylated intermediates of the nascent membrane protein BACE1. EMBO Rep 2008; 9: 916–22.
Kysenius K, Huttunen HJ . Stress-induced upregulation of VLDL receptor alters Wnt-signaling in neurons. Exp Cell Res 2016; 340: 238–47.
Abifadel M, Varret M, Rabès JP, Allard D, Ouguerram K, Devillers M, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet 2003; 34: 154–6.
Allard D, Amsellem S, Abifadel M, Trillard M, Devillers M, Luc G, et al. Novel mutations of the PCSK9 gene cause variable phenotype of autosomal dominant hypercholesterolemia. Hum Mutat 2005; 26: 497–97.
Humphries SE, Cranston T, Allen M, Middleton-Price H, Fernandez MC, Senior V, et al. Mutational analysis in UK patients with a clinical diagnosis of familial hypercholesterolaemia: relationship with plasma lipid traits, heart disease risk and utility in relative tracing. J Mol Med 2006; 84: 203–14.
Maxwell KN, Breslow JL . Proprotein convertase subtilisin kexin 9: the third locus implicated in autosomal dominant hypercholesterolemia. Curr Opin Lipidol 2005; 16: 167–72.
Humphries SE, Whittall RA, Hubbart CS, Maplebeck S, Cooper JA, Soutar AK, et al. Genetic causes of familial hypercholesterolaemia in patients in the UK: relation to plasma lipid levels and coronary heart disease risk. J Med Genet 2006; 43: 943–9.
Maxwell KN, Breslow JL . Adenoviral-mediated expression of Pcsk9 in mice results in a low-density lipoprotein receptor knockout phenotype. Proc Natl Acad Sci U S A 2004; 101: 7100–5.
Park SW, Moon YA, Horton JD . Post-transcriptional regulation of low density lipoprotein receptor protein by proprotein convertase subtilisin/kexin type 9a in mouse liver. J Biol Chem 2004; 279: 50630–8.
Cohen J, Pertsemlidis A, Kotowski IK, Graham R, Garcia CK, Hobbs HH . Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat Genet 2005; 37: 161–5.
Berge KE, Ose L, Leren TP . Missense mutations in the PCSK9 gene are associated with hypocholesterolemia and possibly increased response to statin therapy. Arteriosclerosis Thromb Vasc Biol 2006; 26: 1094–100.
Shan L, Pang L, R, Murgolo N, Lan H, Hedrick J . PCSK9 binds to multiple receptors and can be functionally inhibited by an EGF-A peptide. Biochem Biophys Res Commun 2008; 375: 69–73.
Mcnutt MC, Kwon HJ, Chen C, Chen JR, Horton JD, Lagace TA . Antagonism of secreted PCSK9 increases low density lipoprotein receptor expression in HepG2 cells. J Biol Chem 2009; 284: 10561–70.
Alghamdi RH, O'Reilly P, Lu C, Gomes J, Lagace TA, Basak A . LDL-R promoting activity of peptides derived from human PCSK9 catalytic domain (153–421): design, synthesis and biochemical evaluation. Eur J Med Chem 2015; 92: 890–907.
Palmersmith H, Basak A . Regulatory effects of peptides from the pro and catalytic domains of proprotein convertase subtilisin/kexin 9 (PCSK9) on low-density lipoprotein receptor (LDL-R). Curr Med Chem 2010; 17: 2168–82.
Mitchell T, Chao G, Sitkoff D, Lo F, Monshizadegan H, Meyers D, et al. Pharmacologic profile of the adnectin BMS-962476, a small protein biologic alternative to PCSK9 antibodies for low-density lipoprotein lowering. J Pharmacol Exp Ther 2014; 350: 412–24.
Weider E, Susan-Resiga D, Essalmani R, Hamelin J, Asselin MC, Ashraf Y, et al. Proprotein convertase subtilisin/Kexin type 9 (PCSK9) single domain antibodies are potent inhibitors of LDL receptor degradation. J Biol Chem 2016; 291: 16659–71.
Dahagam C, Goud A, Abdelqader A, Hendrani A, Feinstein MJ, Qamar A, et al. PCSK9 inhibitors and their role in high-risk patients in reducing LDL cholesterol levels: alirocumab. Future Cardiol 2016; 12: 149–57.
Mckenney JM, Koren MJ, Kereiakes DJ, Hanotin C, Ferrand AC, Stein EA . Safety and efficacy of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 serine protease, SAR236553/REGN727, in patients with primary hypercholesterolemia receiving ongoing stable atorvastatin therapy. J Am Coll Cardiol 2012; 59: 2344–53.
Roth EM, Mckenney JM, Hanotin C, Asset G, Stein EA . Atorvastatin with or without an antibody to PCSK9 in primary hypercholesterolemia. N Engl J Med 2012; 367: 1891–900.
Stein EA, Gipe D, Bergeron J, Gaudet D . Effect of a monoclonal antibody to PCSK9, REGN727/SAR236553, to reduce low-density lipoprotein cholesterol in patients with heterozygous familial hypercholesterolaemia on stable statin dose with or without ezetimibe therapy: a phase 2 randomised controlled. Lancet 2012; 380: 29–36.
Kastelein JJP, Robinson JG, Farnier M, Krempf M, Langslet G, Lorenzato C, et al. Efficacy and safety of alirocumab in patients with heterozygous familial hypercholesterolemia not adequately controlled with current lipid-lowering therapy: design and rationale of the ODYSSEY FH studies. Cardiovasc Drugs Ther 2014; 28: 281–9.
Bays H, Gaudet D, Weiss R, Ruiz JL, Watts GF, Gouni-Berthold I, et al. Alirocumab as add-on to atorvastatin versus other lipid treatment strategies: ODYSSEY OPTIONS I randomized trial. J Clin Endocrinol Metab 2015; 100: 3140–8.
Dahagam C, Goud A, Abdelqader A, Hendrani A, Feinstein MJ, Qamar A, et al. PCSK9 inhibitors and their role in high-risk patients in reducing LDL cholesterol levels: evolocumab. Future Cardiol 2016; 12: 139–48.
Cicero AF, Tartagni E, Ertek S . Efficacy and safety profile of evolocumab (AMG145), an injectable inhibitor of the proprotein convertase subtilisin/kexin type 9: the available clinical evidence. Expert Opin Biol Ther 2014; 14: 863–8.
Navarese EP, Kolodziejczak M, Schulze V, Gurbel PA, Tantry U, Lin Y, et al. Effects of proprotein convertase subtilisin/kexin type 9 antibodies in adults with hypercholesterolemia: a systematic review and meta-analysis. Ann Int Med 2015; 163: 40–51.
Winn CL . A 52-week placebo-controlled trial of evolocumab in hyperlipidemia. N Engl J Med 2014; 370: 1809–19.
Cicero AF, Colletti A, Borghi C . Profile of evolocumab and its potential in the treatment of hyperlipidemia. Drug Design Develop Ther 2014; 9: 3073–82.
Gumbiner B, Udata C, Joh T, Liang H, Wan H, Shelton D, et al. Abstract 13322: The effects of single dose administration of RN316 (PF-04950615), a humanized IgG2a monoclonal antibody binding proprotein convertase subtilisin kexin type 9, in hypercholesterolemic subjects treated with and without atorvastatin. Circulation 2012; 126: A13322.
Ballantyne CM, Neutel J, Cropp A, Duggan W, Wang E, Plowchalk D, et al. Efficacy and safety of bococizumab (Rn316/Pf-04950615), a monoclonal antibody against proprotein convertase subtilisin/kexin type 9 in statin-treated hypercholesterolemic subjects: results from a randomized, placebo-controlled, dose-ranging study (Nct: 01592240). J Am Coll Cardiol 2014; 63: A1374.
Sander JD, Joung JK . CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol 2014; 32: 347–55.
Ding Q, Strong A, Patel KM, Ng SL, Gosis BS, Regan SN, et al. Permanent alteration of PCSK9 with in vivo CRISPR-Cas9 genome editing. Circ Res 2014; 115: 488–92.
Wang X, Raghavan A, Chen T, Qiao L, Zhang Y, Ding Q, et al. CRISPR-Cas9 targeting of PCSK9 in human hepatocytes in vivo-brief report. Arteriosclerosis Thromb Vasc Biol 2016; 36: 783–6.
Wilson EM, Bial J, Tarlow B, Bial G, Jensen B, Greiner DL, et al. Extensive double humanization of both liver and hematopoiesis in FRGN mice. Stem Cell Res 2014; 13: 404–12.
Zhang Y, Eigenbrot C, Zhou L, Shia S, Li W, Quan C, et al. Identification of a small peptide that inhibits PCSK9 protein binding to the low density lipoprotein receptor. J Biol Chem 2014; 289: 942–55.
Imanshahidi M, Hosseinzadeh H . Pharmacological and therapeutic effects of Berberis vulgaris and its active constituent, berberine. Phytother Res 2008; 22: 999–1012.
Zhao, GL . Yu, LM, Gao, WL, Duan WX, Jiang B, Liu XD, et al. Berberine protects rat heart from ischemia/reperfusion injury via activating JAK2/STAT3 signaling and attenuating endoplasmic reticulum stress. Acta Pharmacol Sin 2016; 37: 354–67.
Pirillo A, Catapano AL . Berberine, a plant alkaloid with lipid- and glucose-lowering properties: From in vitro evidence to clinical studies. Atherosclerosis 2015; 243: 449–61.
Cameron J, Ranheim T, Kulseth MA, Leren TP, Berge KE . Berberine decreases PCSK9 expression in HepG2 cells. Atherosclerosis 2008; 201: 266–73.
Mendel DB, Crabtree GR . HNF-1, a member of a novel class of dimerizing homeodomain proteins. J Biol Chem 1991; 266: 677–80.
Dong B, Li H, Singh AB, Cao A, Liu J . Inhibition of PCSK9 transcription by berberine involves down-regulation of hepatic HNF1alpha protein expression through the ubiquitin-proteasome degradation pathway. J Biol Chem 2015; 290: 4047–58.
Li H, Dong B, Park SW, Lee HS, Chen W, Liu J . Hepatocyte nuclear factor 1alpha plays a critical role in PCSK9 gene transcription and regulation by the natural hypocholesterolemic compound berberine. J Biol Chem 2009; 284: 28885–95.
Xue H, Ji Y, Wei S, Yu Y, Yan X, Liu S, et al. HGSD attenuates neuronal apoptosis through enhancing neuronal autophagy in the brain of diabetic mice: the role of AMP-activated protein kinase. Life Sci 2016; 153: 23–34.
Jia L, Liu J, Song Z, Pan X, Chen L, Cui X, et al. Berberine suppresses amyloid-beta-induced inflammatory response in microglia by inhibiting nuclear factor-kappaB and mitogen-activated protein kinase signalling pathways. J Pharm Pharmacol 2012; 64: 1510–21.
Wu CY, Tang ZH, Jiang L, Li XF, Jiang ZS, Liu LS . PCSK9 siRNA inhibits HUVEC apoptosis induced by ox-LDL via Bcl/Bax-caspase 9-caspase 3 pathway. Mol Cell Biochem 2012; 359: 347–58.
Tang Z, Jiang L, Peng J, Ren Z, Wei D, Wu C, et al. PCSK9 siRNA suppresses the inflammatory response induced by oxLDL through inhibition of NF-κB activation in THP-1-derived macrophages. Int J Mol Med 2012; 30: 931–8.
Sultana N, Ata A . Oleanolic acid and related derivatives as medicinally important compounds. J Enzyme Inhibit Med Chem 2008; 23: 739–56.
Ovesná Z, Vachálková A, Horváthová K, Tóthová D . Pentacyclic triterpenoic acids: new chemoprotective compounds. Minireview. Neoplasma 2004; 51: 327–33.
Kim NY, Lee MK, Park MJ, Kim SJ, Park HJ, Choi JW, et al. Momordin Ic and oleanolic acid from Kochiae Fructus reduce carbon tetrachloride-induced hepatotoxicity in rats. J Med Food 2005; 8: 177–83.
Jie LI, Guo WJ, Yang QY . Effects of ursolic acid and oleanolic acid on human colon carcinoma cell line HCT15. World J Gastroenterol 2002; 8: 493–5.
Tsai SJ, Yin MC . Antioxidative and anti-inflammatory protection of oleanolic acid and ursolic acid in PC12 cells. J Food Sci 2008; 73: H174–H78.
Rodriguez-Rodriguez R, Stankevicius E, Herrera MD, Ostergaard L, Andersen MR, Ruiz-Gutierrez V, et al. Oleanolic acid induces relaxation and calcium-independent release of endothelium-derived nitric oxide. Br J Pharmacol 2008; 155: 535–46.
Xin W, Rui L, Wei Z, Zhang X, Liao N, Zhao W, et al. Oleanolic acid improves hepatic insulin resistance via antioxidant, hypolipidemic and anti-inflammatory effects. Mol Cell Endocrinol 2013; 376: 70–80.
Li XF, Jiang L, Fang CM, Ren Z, Tang ZH, Liu LS . Effects of four Chinese medicine monomers on expression of PCSK9 in HepG2 cells in vitro. Chin J Arteriosclerosis 2011; 19: 264.
Sehgal A, Vaishnaw A, Fitzgerald K . Liver as a target for oligonucleotide therapeutics. J Hepatol 2013; 59: 1354–9.
Graham MJ, Lemonidis KM, Whipple CP, Subramaniam A, Monia BP, Crooke ST, et al. Antisense inhibition of proprotein convertase subtilisin/kexin type 9 reduces serum LDL in hyperlipidemic mice. J Lipid Res 2007; 48: 763–7.
Kauppinen S, Vester B, Wengel J . Locked nucleic acid (LNA): High affinity targeting of RNA for diagnostics and therapeutics. Drug Discov Today Technol 2005; 2: 287–90.
Frank-Kamenetsky M, Grefhorst A, Anderson NN, Racie TS, Bramlage B, Akinc A, et al. Therapeutic RNAi targeting PCSK9 acutely lowers plasma cholesterol in rodents and LDL cholesterol in nonhuman primates. Proc Natl Acad Sci U S A 2008; 105: 11915–20.
Fitzgerald K, Frank-Kamenetsky M, Shulga-Morskaya S, Liebow A, Bettencourt BR, Sutherland JE, et al. Effect of an RNA interference drug on the synthesis of proprotein convertase subtilisin/kexin type 9 (PCSK9) and the concentration of serum LDL cholesterol in healthy volunteers: a randomised, single-blind, placebo-controlled, phase 1 trial. Lancet 2014; 383: 60–8.
Gustafsen C, Kjolby M, Nyegaard M, Mattheisen M, Lundhede J, Buttenschøn H, et al. The hypercholesterolemia-risk gene SORT1 facilitates PCSK9 secretion. Cell Metab 2014; 19: 310–8.
Chen XW, Wang H, Bajaj K, Zhang P, Meng ZX, Ma D, et al. SEC24A deficiency lowers plasma cholesterol through reduced PCSK9 secretion. Elife 2013; 2: e00444.
Miranda MX, Tits LJV, Lohmann C, Arsiwala T, Winnik S, Tailleux A, et al. The Sirt1 activator SRT3025 provides atheroprotection in ApoE−/− mice by reducing hepatic Pcsk9 secretion and enhancing Ldlr expression. Eur Heart J 2015; 36: 51–9.
Chorba JS, Shokat KM . The proprotein convertase subtilisin/kexin type 9 (PCSK9) active site and cleavage sequence differentially regulate protein secretion from proteolysis. J Biol Chem 2014; 289: 29030–43.
Carare RO, Teeling JL, Hawkes CA, Püntener U, Weller RO, Nicoll JA, et al. Immune complex formation impairs the elimination of solutes from the brain: implications for immunotherapy in Alzheimer's disease. Acta Neuropathol Commun 2013; 1: 1–11.
Merega E, Prisco SD, Severi P, Kalfas F, Pittaluga A . Antibody/receptor protein immunocomplex in human and mouse cortical nerve endings amplifies complement-induced glutamate release. Neurosci Lett 2015; 600: 50–5.
Koren MJ, Giugliano RP, Raal FJ, Sullivan D, Bolognese M, Langslet G, et al. Efficacy and safety of longer-term administration of evolocumab (AMG 145) in patients with hypercholesterolemia: 52-week results from the open-label study of long-term evaluation against LDL-C (OSLER) randomized trial. Circulation 2014; 129: 234–43.
Cicero AF, Tartagni E, Ertek S . Safety and tolerability of injectable lipid-lowering drugs: a review of available clinical data. Expert Opin Drug Safety 2014; 13: 1–8.
Drewe E, Powell RJ . Clinically useful monoclonal antibodies in treatment. J Clin Pathol 2002; 55: 81–5.
Geeganage C, Wilcox R, Bath PM . Triple antiplatelet therapy for preventing vascular events: a systematic review and meta-analysis. BMC Med 2010; 8: 1–14.
Schmitt C, Abt M, Ciorciaro C, Kling D, Jamois C, Schick E, et al. First-in-man study with inclacumab, a human monoclonal antibody against P-selectin. J Cardiovasc Pharmacol 2015; 65: 611–9.
Zhang TT, Li W, Meng G, Wang P, Liao W . Strategies for transporting nanoparticles across the blood-brain barrier. Biomater Sci 2016; 4: 219–29.
Kazi DS, Moran AE, Coxson PG, Penko J, Ollendorf DA, Pearson SD, et al. Cost-effectiveness of PCSK9 inhibitor therapy in patients with heterozygous familial hypercholesterolemia or atherosclerotic cardiovascular disease. JAMA 2016; 316: 743–53.
Wang X, Raghavan A, Chen T, Qiao L, Zhang Y, Ding Q, et al. CRISPR-Cas9 targeting of PCSK9 in human hepatocytes in vivo — brief report. Arterioscler Thromb Vasc Biol 2016; 36: 783–6.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (No 81200217, 81370376), the Natural Science Foundation of Hunan province (No 2015JJ4097), the Key Project of Science and Technology Department of Hu-nan Province (No 2015JC3081), the Key Project of the Educational Department of Hu-nan Province (No 15A137), the Visiting Scholar Foundation of Key Laboratory of Biorheological Science and Technology (Chongqing University), the Ministry of Education (No CQKLBST-2014-002, CQKLBST-2015-004), and the Construct Program of the Key Discipline in Hu-nan Province.
Author information
Authors and Affiliations
Corresponding authors
PowerPoint slides
Rights and permissions
About this article

Cite this article
He, Ny., Li, Q., Wu, Cy. et al. Lowering serum lipids via PCSK9-targeting drugs: current advances and future perspectives. Acta Pharmacol Sin 38, 301–311 (2017). https://doi.org/10.1038/aps.2016.134
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2016.134
Keywords
This article is cited by
-
Impact of Lowering Low-Density Lipoprotein Cholesterol with Contemporary Lipid-Lowering Medicines on Cognitive Function: A Systematic Review and Meta-Analysis
Cardiovascular Drugs and Therapy (2021)
-
Relationship between Lipoprotein (a) and cognitive function – Results from the Berlin Aging Study II
Scientific Reports (2020)
-
Single-Strand DNA-Like Oligonucleotide Aptamer Against Proprotein Convertase Subtilisin/Kexin 9 Using CE-SELEX: PCSK9 Targeting Selection
Cardiovascular Drugs and Therapy (2020)
-
RNA-guided transcriptional silencing in vivo with S. aureus CRISPR-Cas9 repressors
Nature Communications (2018)
