
Abstract
Alzheimer's disease (AD) is a progressive neurodegenerative disease leading to the irreversible loss of brain neurons and cognitive abilities, and the vicious interplay between oxidative stress (OS) and tauopathy is believed to be one of the major players in AD development. Here, we demonstrated the capability of the small molecule N-(1,3-benzodioxol-5-yl)-2-[5-chloro-2-methoxy(phenylsulfonyl)anilino]acetamide (LX2343) to ameliorate the cognitive dysfunction of AD model rats by inhibiting OS-induced neuronal apoptosis and tauopathy. Streptozotocin (STZ) was used to induce OS in neuronal cells in vitro and in AD model rats that were made by intracerebroventricular injection of STZ (3 mg/kg, bilaterally), and Morris water maze test was used to evaluate the cognitive dysfunction in ICV-STZ rats. Treatment with LX2343 (5–20 μmol/L) significantly attenuated STZ-induced apoptosis in SH-SY5Y cells and mouse primary cortical neurons by alleviating OS and inhibiting the JNK/p38 and pro-apoptotic pathways. LX2343 was able to restore the integrity of mitochondrial function and morphology, increase ATP biosynthesis, and reduce ROS accumulation in the neuronal cells. In addition, LX2343 was found to be a non-ATP competitive GSK-3β inhibitor with IC50 of 1.84±0.07 μmol/L, and it potently inhibited tau hyperphosphorylation in the neuronal cells. In ICV-STZ rats, administration of LX2343 (7, 21 mg·kg−1·d−1, ip, for 5 weeks) efficiently improved their cognitive deficits. LX2343 ameliorates the cognitive dysfunction in the AD model rats by suppressing OS-induced neuronal apoptosis and tauopathy, thus highlighting the potential of LX2343 for the treatment of AD.
Similar content being viewed by others


Introduction
Alzheimer's disease (AD) is a progressive neurodegenerative disease characterized by memory and cognitive declines, disorientation, and personality changes, and eventually leads to death1. Currently, more than 35 million people are suffering from AD, and this figure is projected to grow to 106.8 million people by 20502. Apart from the slow and progressive degeneration process with synaptic loss leading to a final neuronal death, the accumulation of amyloid β (Aβ) plaques and the formation of neurofibrillary tangles (NFTs) in the brain are accepted as the main hallmarks of AD3. For a long time, there has been considerable effort devoted to searching for medications that can be used to treat AD. However, to date, an effective treatment for this disease has not been found because of its complicated pathogenesis, as indicated by the fact that a series of potential anti-AD drugs had favorable therapeutic effects in vitro but failed to work in patients4. Therefore, a new strategy is urgently needed for the discovery of anti-AD drugs.
Studies have demonstrated that oxidative imbalance is a manifestation of AD that even precedes Aβ deposition and NFTs formation, and plays a crucial role in neuronal degeneration5,6. Increased oxidative stress (OS) has been implicated in the aging process and is a result of the imbalance between the generation and clearance of reactive oxygen species (ROS). As a main source of ROS, progressive mitochondrial dysfunction may in turn amplify oxidative deficits. The brain is highly vulnerable to oxidative imbalance due to its intensive energy demand, high oxygen consumption, and relative paucity of antioxidants and related enzymes. Thus, it is no wonder that OS-mediated damage is extensively observed in AD. Since mitochondria lie in the center of intrinsic apoptotic events, persistent OS and mitochondrial dysfunction likely lead to further activation of neuronal apoptosis cascades and deficits of cognitive and behavioral functions7. Interestingly, evidence has revealed that OS is closely linked to tau pathology, including tau hyperphosphorylation, polymerization and toxicity7.
Tau protein is associated with microtubule dynamics, which are mainly responsible for maintaining neuronal morphology and functional integrity8. Abnormal hyperphosphorylation of tau impairs its capacity for binding to microtubules and maintaining microtubule assembly, resulting in microtubule disorganization, transport defects along axonal microtubules and self-aggregation into NFTs, causing further toxic effects in neuronal cells9. In addition, accumulation of hyperphosphorylated tau promotes ROS production, while ROS in turn directly stimulates tau hyperphosphorylation and aggregation. Such a vicious cycle largely propagates the pathogenesis of AD, thereby accelerating the progression of this disease7.
In our previous work10, we reported that the small molecular compound N-(1,3-benzodioxol-5-yl)-2-[5-chloro-2-methoxy(phenylsulfonyl)anilino]acetamide (LX2343, Figure 1A) was able to alleviate Aβ accumulation by both promoting its clearance and suppressing its production under streptozotocin (STZ)-induced AD pathological conditions and effectively ameliorates cognitive dysfunction in APP/PS1 transgenic mice. Given that oxidative damage and tau hyperphosphorylation are two of the characteristic pathological features induced by STZ in vitro and in vivo11, here, we performed the related assays in an attempt to investigate the potential regulation of OS-involved events and tau hyperphosphorylation by LX2343 under STZ-induced pathological conditions. Interestingly, we discovered that LX2343 effectively reversed the STZ-induced neuronal apoptosis and tau hyperphosphorylation in vitro and in vivo, and this neuroprotective effect of LX2343 was ascribed to its function in the inhibition of OS and tauopathy. Combined with our previous discovery of the Aβ inhibitory efficacy of this agent10, our results demonstrate that LX2343 is a multi-target agent that exhibits a high capability for ameliorating multi-abnormalities of AD pathogenesis, thus strongly supporting the potential of LX2343 in the treatment of AD.

LX2343 protected neurons from damage. (A) Structure of LX2343. (B, C) STZ induced cell death in SH-SY5Y and primary neuronal cells (MTT assay) (one-way ANOVA, Dunnett's multiple comparison test. P<0.01 versus DMSO). (D, E) LX2343 reduced STZ-induced cell death in SH-SY5Y and primary neuronal cells (MTT assay) (one-way ANOVA, Dunnett's multiple comparison test. *P<0.05, **P<0.01 versus STZ. ##P<0.01 versus DMSO). (F) SH-SY5Y cells were stained with Annexin V-FITC, and Annexin V-FITC positive cells were determined by FACS (one-way ANOVA, Dunnett's multiple comparison test. **P<0.01 versus STZ. ##P<0.01 versus DMSO). (G, H) Western blot and quantification results demonstrated that LX2343 ameliorated the STZ-induced suppression of PSD95, synaptophysin, and VAMP2 (one-way ANOVA, Dunnett's multiple comparison test. *P<0.05, **P<0.01 versus STZ. ##P<0.01 versus DMSO). All values were presented as the means±sem (n=3). GAPDH was used as the loading control in Western blot assays. Data were obtained from three independent experiments.
Materials and methods
Materials
All cell culture reagents were purchased from Gibco (Invitrogen, Grand Island, NY, USA). 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), streptozotocin (STZ), vitamin E (VE), and lithium chloride were purchased from Sigma-Aldrich (St Louis, MO, USA). SB216763 and PHA-793887 were purchased from Selleck (Houston, TX, USA). LX2343 was purchased from the commercial compound library (SPECS, Zoetermeer, Netherlands).
Cell culture
SH-SY5Y cells were grown in a 1:1 mixture of Dulbecco's modified Eagle medium and Ham's F-12 (DMEM/F12) supplemented with 10% fetal bovine serum (FBS) and 100 units/mL of penicillin-streptomycin. Cells were cultured in a humidified incubator with 5% CO2 at 37 °C.
Primary cortical neuron culture
Primary neuronal cultures from embryonic mouse brains (embryonic d 16 to 18) were created according to the approach described in the literature12. Briefly, cerebral cortices were separated from the brain, minced into small pieces, digested with D-Hanks buffer (5.4 mmol/L KCl, 0.41 mmol/L KH2PO4, 138 mmol/L NaCl, 4.5 mmol/L NaHCO3, 0.22 mmol/L Na2HPO4, pH 7.4) containing 0.125% trypsin and 200 U/mL DNase (Sigma-Aldrich, St Louis, MO, USA) and then incubated for 15 min at 37 °C, followed by mechanical dissociation by pipetting in DMEM with 10% FBS. The isolated neurons were seeded at a density of 500 000 cells/mL on poly-D-lysine (Sigma-Aldrich, St Louis, MO, USA)-coated cell culture plates. After 6 h, the medium was replaced by a neurobasal medium supplemented with 2% B27, 0.5 mmol/L L-glutamine, and 50 U/mL penicillin-streptomycin. Cultures were kept in a humidified incubator with 5% CO2 at 37 °C.
MTT assay
The viability of the cultured cells was determined by assaying the reduction of MTT to formazan. SH-SY5Y or neuronal cells were seeded overnight in 48-well plates at a density of 105 cells/well in 100 μL medium. The cells were then incubated with different concentrations of LX2343 (5, 10, or 20 μmol/L) and STZ (0.8 mmol/L for SH-SY5Y cells, 0.4 mmol/L for neuronal cells) for 24 h and washed twice with PBS, followed by the addition of MTT (0.5 mg/mL). After incubation at 37 °C for 4 h, 200 μL DMSO was added to dissolve the formazan crystals, and the absorbance at 490 nm was measured using an M5 spectrophotometer (Molecular Devices, Sunnyvale, CA, USA).
FACS assay
SH-SY5Y cells were incubated with STZ (0.8 mmol/L) for 24 h and then collected. Cell apoptosis was measured using an Annexin V-FITC apoptosis detection kit (Beyotime Company, China) following the manufacturer's protocol. The proportion of cell apoptosis was determined by Flow Cytometry (BD Bioscience Company, San Jose, CA, USA).
Intracellular ROS assay
The production of intracellular ROS was measured by fluorescence microscopy (Olympus Company, Tokyo, Japan) using an oxidation-sensitive probe, 20,70-dichlorofluorescein diacetate (DCFH-DA) (Beyotime Company, Haimen, China). After exposing either the SH-SY5Y or the primary neuronal cells to STZ (0.8 mmol/L or 0.4 mmol/L, respectively) with different concentrations of LX2343 (5, 10, or 20 μmol/L) for 8 h, the ROS level was measured according to the manufacturer's protocol.
Mitochondrial membrane potential assay
The mitochondrial membrane potential (MMP) was determined using the MMP Kit (Beyotime Company, Haimen, China). The assay involved exposing either SH-SY5Y or neuronal cells to STZ (0.8 or 0.4 mmol/L, respectively) with different concentrations of LX2343 (5, 10, or 20 μmol/L) for 8 h in 12-well plates. The assay details were as described in the manufacturer's protocol.
Mitochondrial function assay
The concentration of ATP was measured using the ATP assay kit (Beyotime Company, Haimen, China). The assay involved exposing either SH-SY5Y or neuronal cells to STZ (0.8 or 0.4 μmol/L, respectively) with different concentrations of LX2343 (5, 10, or 20 μmol/L) for 8 h in 12-well plates. The assay details were as described in the manufacturer's protocol.
Transmission electron microscopy-based assay
After incubation with STZ (0.8 mmol/L) or LX2343 (20 μmol/L), SH-SY5Y cells were fixed with 2.5% glutaraldehyde and 2% paraformaldehyde in 0.1 mol/L sodium phosphate buffer, pH 7.2, for 2 h. The cells were collected, pelleted, washed with sodium phosphate buffer, and post-fixed with 2% osmium tetroxide for 2 h at 4 °C. The pellets were dehydrated in increasing concentrations of ethanol and then embedded. Thin sections (70 nm) were cut with a Leica EM UC7 ultramicrotome (Leica Microsystems, Wetzlar, Germany) and stained with uranyl acetate and lead acetate. The sections were observed using a Tecnai G2 Spirit transmission electron microscope (FEI, Eindhoven, Netherlands).
GSK-3β enzymatic activity assay
GSK-3β inhibition by LX2343 was evaluated by performing an ELISA according to the published approach13. Briefly, recombinant human GSK-3β (Invitrogen, Grand Island, NY, USA) was added to the reactions with the substrate-recombinant human TAU-441 (Merck Millipore, Darmstadt, Germany) in a 5:1 ratio. The reaction buffer contained 40 mmol/L HEPES, 5 mmol/L MgCl2, 1 mmol/L EDTA, 50 mg/mL of heparin and 100, 30, or 10 μmol/L ATP, and had a pH of 7.2. The reaction mixture was incubated at 37 °C for 1 h. The amount of phosphorylated TAU-441 was measured using a human Tau [pS396] ELISA kit (Invitrogen, Grand Island, NY, USA).
Western blot
The cell-based assays involved exposing either the SH-SY5Y or the primary neuronal cells to STZ (0.8 mmol/L or 0.4 mmol/L, respectively), treating with different concentrations of LX2343 (5, 10, or 20 μmol/L), and then lysing with RIPA buffer (Thermo Scientific, Waltham, MA, USA) containing a protease inhibitor cocktail (Thermo Scientific, Waltham, MA, USA). The protein concentration was determined using a BCA protein assay kit (Thermo Scientific, Waltham, MA, USA). Proteins were mixed with 2× SDS-PAGE sample buffer (25% SDS, 62.5 mmol/L Tris-HCl, pH 6.8, 25% glycerol, 0.5 mol/L DTT, and 0.1% bromophenol blue) and then boiled for 15 min at 99 °C.
For cytochrome c (cyt c) analysis, both cytoplasmic and mitochondrial extracts were collected separately using the Mitochondria/Cytosol Fraction kit (Sangon Biotech, Shanghai, China). The cytoplasmic protein concentration was determined using the BCA protein assay kit (Thermo Scientific, Waltham, MA, USA). The mitochondrial protein concentration was determined using the Non-Interference Protein Assay Kit (Sangon Biotech, Shanghai, China), and mitochondrial proteins were mixed with 5× SDS-PAGE sample buffer (10% SDS, 250 mmol/L Tris-HCl, pH 6.8, 50% glycerol, 0.5 mol/L DTT, and 0.25% bromophenol blue) and then boiled for 15 min at 99 °C.
The brain tissue-based assays involved homogenizing the rat brain tissue with RIPA buffer (Thermo Scientific, Waltham, MA, USA) containing a protease inhibitor cocktail and phosphatase inhibitor cocktails (Thermo Scientific, Waltham, MA, USA) using a hand-held motor. The homogenates were kept on ice for 1 h to ensure that the cells lysed completely. The homogenates were then centrifuged at 20 000×g for 30 min at 4 °C. The supernatant was collected and the protein concentration was determined using the BCA protein assay kit. Equal amounts of lysate (4 mg/mL protein) were mixed with 2× SDS-PAGE sample buffers and then boiled for 15 min at 99 °C. Three individual samples of each of the four groups are shown in Results because only 12 lanes were available in each gel; the other samples were analyzed but are not shown in the figures.
Antibodies against p-JNK, JNK, p-p38, p38, p-c-Jun, c-Jun, p-Erk, Erk, cleaved caspase-3, caspase-3, Bcl-2, Bcl-xl, BAX, p-Bad, Bad, PSD95, Synaptophysin, VAMP2, Tau, P9-GSK-3β, p25, p35, CDK5, and GAPDH were obtained from Cell Signaling Technology (Danvers, MA, USA). An antibody against P216-GSK-3β was purchased from Santa Cruz (TX, USA). Antibodies against P396-Tau, P199-Tau, and P231-Tau were purchased from Abcam (Cambridge, UK). For Western blot analysis, cell or tissue extracts were separated by SDS-PAGE and transferred to a polyvinylidene difluoride membrane filter (GE Healthcare, Madison, WI, USA). After incubation with the corresponding antibodies overnight, the blots were visualized using the Dura detection system (Thermo Scientific, Waltham, MA, USA).
Animal experiments
All animal experiments were performed according to the institutional ethical guidelines on animal care of Shanghai Institute of Materia Medica, Chinese Academy of Sciences.
Forty adult male Sprague Dawley (SD) rats weighing 140–160 g were used in the study. The rats were randomly divided into four groups (ten in each group) and maintained under standard conditions at room temperature (22 °C) with a 12 h light/dark cycle. The animals were purchased from Slac Laboratory Animal Co Ltd (Shanghai, China). The rats in the first group (V) treated with vehicle did not have surgery because previous studies14 and our results (Supplementary Figure S1A-S1B) both demonstrated that there was no significant difference in the behavior and cognitive function of control rats (without surgery) and that of sham rats (recipients of an ICV injection of vehicle). The rats in the second group (SV) were injected with ICV-STZ (3 mg/kg bilaterally) and vehicle, and the rats in the third (SL) and fourth (SH) groups were injected with ICV-STZ and different doses of LX2343 (7 and 21 mg/kg, respectively). LX2343 was dissolved in 3% DMSO and 5% Tween-80. Before surgery, pre-administration of LX2343 was carried out for 2 weeks. The treatment was maintained for 21 consecutive days after surgery. The brief surgical procedure was performed as follows. Rats were anesthetized with 10% chloral hydrate intraperitoneally and placed on a stereotaxic frame. Holes were drilled in the skull on both sides over the lateral ventricles according to the following coordinates: 0.8 mm posterior to the bregma, 1.5 mm lateral to the saggital suture, and 3.6 mm beneath the surface of the brain. The lesion groups received a bilateral ICV injection of STZ (3 mg/kg, 10 μL/each side). STZ was dissolved in artificial CSF (147 mmol/L NaCl, 2.9 mmol/L KCl, 1.6 mmol/L MgCl2, 1.7 mmol/L CaCl2, and 2.2 mmol/L dextrose). All microinjections were performed slowly at a speed of 2 μL/min, and the needle remained in position for more than 2 min to prevent reflux along the injection tract.
Morris water maze test
The Morris water maze (MWM) test was carried out as previously described15. Briefly, in the training trials, rats were trained to find the invisible submerged white platform in a circular pool (120 cm in diameter, 50 cm deep) filled with ink-tinted water using a variety of visual cues located on the pool wall. Three trials were performed each day for 5 consecutive days (the first 4 d were trials for the latency assay and the fifth day for the crossing-times assay). In each trial, rats were given 60 s to find the invisible platform. The animals were allowed to stay at the platform for 10 s if they found the platform within the given time. However, if the animals failed to find the platform within the given time, they were manually placed on the platform and left there for 10 s. On the last day, after the training trials, a probe trial was performed by removing the platform. The animals were allowed to swim for 60 s in search of the platform. All data were collected for animal performance analysis. For data analysis, the pool was divided into four equal quadrants, formed by imaging lines that intersected in the center of the pool at right angles, called north, south, east, and west.
Immunohistochemistry
Immunohistochemistry assays were performed to detect the expression of P396-Tau protein. The tissue was embedded in paraffin and sectioned with a microtome to create sections 5-μm thick. The slides were dewaxed and washed with PBS several times and preincubated in 1% horse serum for 45 min at 37 °C. Thereafter, the slides were incubated overnight at 4 °C with the primary antibody anti-P396-Tau polyclonal (dilution 1:400). Then, the sections were incubated with fluorescent secondary detection reagents (1:500, Invitrogen, Grand Island, NY, USA). The slides were observed under a fluorescence microscope.
TUNEL assay
Cell death in animal brain tissue was detected using the in situ cell death detection kit, POD (Roche, Rotkreuz, Switzerland), according to the manufacturer's protocol. Briefly, the assay was based on the detection of DNA fragmentation resulting from apoptosis signaling cascades by labeling the DNA strand breaks using terminal deoxynucleotidyl transferase16.
Statistical analysis
All data were obtained from three independent experiments in which experiments were performed in triplicate, at least. All data are presented as the mean±SEM, and the exact values of the mean±SEM are presented in Supplementary Table S1. The significant difference between multiple treatments and the control was analyzed using a one-way ANOVA with Dunnett's post-test. The STZ treatment (SV) was used as the positive control to evaluate the ameliorative effects of compounds on AD pathologies. A P value of less than 0.05 was considered to be statistically significant.
Results
LX2343 protected neurons from apoptosis by ameliorating OS and mitochondrial dysfunction
LX2343 protected neurons from apoptosis
Given that STZ has been widely used to induce OS-mediated cell apoptosis both in vitro and in vivo11,17, we investigated the potential neuronal protective effects of LX2343 in SH-SY5Y and primary neuronal cells by using STZ as an inducer for cell death. The effect of STZ on neuron viability was first evaluated using the MTT assay. The results indicated that 0.4–3.2 mmol/L STZ showed a dose-dependent inhibitory activity on cell viability (Figure 1B, 1C) and, therefore, the STZ dose was set at 0.8 mmol/L for SH-SY5Y and 0.4 mmol/L for primary neuronal cells in all subsequent assays. As demonstrated in Figure 1D and 1E, STZ treatment induced an 18.11%±1.15% and 27.82%±0.82% decrease in cell viability for SH-SY5Y and primary neuronal cells, respectively, while co-incubation with LX2343 effectively antagonized the effects of STZ. In addition, quantitative evaluation of apoptosis in SH-SY5Y cells using Annexin V-FITC staining further demonstrated the capability of LX2343 to prevent STZ-induced apoptosis (Figure 1F). Compared with the controls, the apoptosis rate of the SH-SY5Y cells was increased to 21.88%±0.91% when the cells were stimulated with STZ, and the apoptosis rate was decreased to 14.57%±0.66% after the cells were co-incubated with 20 μmol/L LX2343. These data indicated that treating SH-SY5Y and primary neuronal cells with LX2343 could reduce cell death and increase cell viability.
Next, given that STZ-induced OS stimulation may disturb synapse integrity18, we examined whether LX2343 could also regulate the expression of synapse-associated proteins in primary neuronal cells. For this purpose, the protein levels of the postsynaptic density protein 95 (PSD95), synaptophysin, and vesicle-associated membrane protein 2 (VAMP2), which are crucial for neurotransmission and synaptic plasticity19, were detected by Western blot. As expected, compared with the controls, the expression of PSD95, synaptophysin, and VAMP2 was decreased to 66%±3.7%, 46%±4.7%, and 50%±4.1%, respectively, after exposure to STZ for 8 h. However, LX2343 dose-dependently abolished the STZ-induced reduction in the expression of these proteins, thereby implying the potential protection of this compound against synapse integrity (Figure 1G, 1H).
Taken together, all these results indicated that LX2343 exhibited neuronal protective effects.
LX2343 alleviated STZ-induced OS and ameliorated mitochondrial dysfunction
Because ROS and mitochondrial dysfunction are two of the primary pathological features of AD20 and are widely implicated in STZ-induced cell death21, we next examined whether LX2343 affected ROS accumulation in cells with antioxidant VE as a positive control22. As expected, ROS generation was greatly increased after STZ incubation in both SH-SY5Y (162.45%±0.043%) (Figure 2A, 2B) and primary neuronal cells (126.35%±3.50%) (Figure 2C, 2D), whereas 100 μmol/L VE inhibited such STZ-induced effects. Furthermore, LX2343 treatment efficiently inhibited the accumulation of intracellular ROS in both SH-SY5Y (Figure 2A, 2B) and primary neuronal cells (Figure 2C, 2D). Moreover, the combination of LX2343 with VE exhibited a synergistic action in the prevention of STZ-induced apoptosis (Supplementary Figure S2). These results indicated that the neuroprotective ability of LX2343 involves an inhibitory effect on ROS accumulation.

LX2343 alleviated STZ-induced OS and ameliorated mitochondrial dysfunction. (A–D) Decreased ROS levels in LX2343-treated SH-SY5Y and primary neuronal cells. DCFH-DA fluorescence assayed by fluorescence and fluorimetry (one-way ANOVA, Dunnett's multiple comparison test. *P<0.05, **P<0.01 versus STZ. ##P<0.01 versus DMSO), scale bar: 100 μm. (E, F) Cells were stained with JC-1, and the mitochondrial membrane potential was detected (Δψ=525:590/490:530; one-way ANOVA, Dunnett's multiple comparison test. **P<0.01 versus STZ. ##P<0.01 versus DMSO). (G) Transmission electron microscopy images of mitochondria in SH-SY5Y cells, n=4, scale bar: 0.5 μm. (H, I) The intracellular ATP level was detected using a microplate reader (one-way ANOVA, Dunnett's multiple comparison test. **P<0.01 versus STZ. ##P<0.01 versus DMSO). All values were presented as the means±sem (n=3). Data were obtained from three independent experiments.
According to previous reports, mitochondria are major producers of ROS, playing a fundamental role in the maintenance of internal homeostasis, and alterations in mitochondrial membrane integrity are one of the crucial events in ROS generation and early apoptosis23. Therefore, the effect of LX2343 on mitochondrial membrane potential (MMP) in either SH-SY5Y or primary neuronal cells was evaluated using a JC-1 detection kit. In cultures exposed to STZ, the MMP of SH-SY5Y and primary neuronal cells dropped to 80.18%±1.08% and 88.67%±0.32%, respectively. LX2343 clearly antagonized the STZ-induced decrease in MMP (Figure 2E, 2F), implying that LX2343 could help to maintain mitochondrial membrane integrity. In addition, the results of the assay of mitochondrial ultrastructure morphology in SH-SY5Y cells also demonstrated that the cells that received the STZ treatment had swollen mitochondria with decreased matrix electron density and disorganized and fragmented cristae, while LX2343 incubation effectively ameliorated these STZ-induced abnormalities of mitochondria (Figure 2G).
Moreover, given that mitochondrial dysfunction may promote ROS production due to an impairment in normal regulation of the redox state and energy metabolism, the level of intracellular ATP was measured to evaluate mitochondrial function. As expected, the ATP content of the SH-SY5Y and primary neuronal cells decreased to 79.87%±2.048% and 80.25%±1.53%, respectively, when treated with STZ, but co-treatment of the SH-SY5Y and primary neuronal cells with LX2343 effectively attenuated the reduction of ATP in a dose-dependent manner (Figure 2H, 2I).
Therefore, all the data suggested that LX2343 decreased the STZ-induced OS and ameliorated mitochondrial dysfunction.
LX2343 inhibited JNK/p38 signaling
Since intracellular ROS activate the mitogen-activated protein kinase (MAPK) pathway in the mediation of STZ-induced cell death24,25, we next investigated whether LX2343 has a regulatory effect on the MAPK pathway. The results demonstrated that incubating SH-SY5Y and primary neuronal cells with STZ for 4 h induced an ∼2.5-fold increase in JNK and p38 phosphorylation compared with the controls (Figure 3A), whereas LX2343 treatment antagonized the STZ-induced increase in phosphorylated JNK, c-Jun, and p38 in both SH-SY5Y (Figure 3B, 3C) and primary neuronal cells (Figure 3D, 3E) but had no effects on ERK (Figure 3A and 3F) or p53 (Figure 3F) in SH-SY5Y cells. These data thus indicated that the neuronal protective effect of LX2343 involves JNK/p38 pathway inhibition.

LX2343 inhibited JNK/p38 signaling pathway. (A) Western blot results for phosphorylation of JNK, p38, and ERK in SH-SY5Y cell lysates indicated that STZ incubation induced a time-dependent increase in JNK and p38 phosphorylation. (B–E) Western blot and quantification results demonstrated that LX2343 reduced JNK, c-Jun, and p38 phosphorylation in SH-SY5Y and primary neuronal cells (one-way ANOVA, Dunnett's multiple comparison test. *P<0.05, **P<0.01 versus STZ. #P<0.05, ##P<0.01 versus DMSO). (F) Western blot results demonstrated that LX2343 had no effect on ERK or p53 in SH-SY5Y cells. All values were presented as the means±sem (n=3). GAPDH was used as the loading control in Western blot assays. Data were obtained from three independent experiments.
LX2343 regulated apoptosis-relevant proteins
The Bcl-2 family consists of both anti- and pro-apoptotic members that share four conserved regions known as Bcl-2 homology domains (BH1-4). The relative amounts of anti- and pro-apoptotic proteins determine the fate of cells, whether to remain viable or to enter into apoptosis26. Dysregulation of Bcl-2 family members damages mitochondrial outer membrane permeability and promotes efflux of cytochrome c and other death-inducing factors from mitochondria to initiate the apoptotic caspase cascade27. Based on these facts, we investigated the regulation of Bcl-2 family proteins by LX2343 in both SH-SY5Y and primary neuronal cells using Western blot in an attempt to explore the mechanism underlying the protective effects of LX2343 against STZ-induced neurotoxicity. The results indicated that STZ treatment decreased the expression levels of the anti-apoptotic proteins Bcl-2 and Bcl-xl, increased the expression levels of the pro-apoptotic protein Bax, and reduced the phosphorylation of Bad, which self-inactivates Bad activity, while LX2343 incubation inhibited the apoptotic process by up-regulating anti-apoptotic proteins and down-regulating pro-apoptotic proteins (Figure 4A–4D). Similarly, LX2343 ameliorated the STZ-induced increase in cleaved caspase-3 (Figure 4A–4D). In addition, the regulation of cyt c distribution by LX2343 was also confirmed with cytosolic and mitochondrial fractions prepared using SH-SY5Y cells; here, LX2343 treatment markedly abolished STZ-induced cytochrome c release (Figure 4E, 4F).

LX2343 regulated the apoptosis-relevant proteins. (A–D) Western blot and quantification results demonstrated that LX2343 increased Bcl-2 or Bcl-xl protein level, increased Bad phosphorylation, decreased Bax protein level, and inhibited caspase 3 cleavage in SH-SY5Y and primary neuronal cells (one-way ANOVA, Dunnett's multiple comparison test, *P<0.05, **P<0.01 versus STZ, #P<0.05, ##P<0.01 versus DMSO). (E, F) Cytosolic and mitochondrial fraction were prepared, and cyt c distribution was analyzed (one-way ANOVA, Dunnett's multiple comparison test, *P<0.05 versus STZ, #P<0.05, ##P<0.01 versus DMSO). All values were presented as mean±SEM (n=3). GAPDH was used as loading control in Western blot assays. Data were obtained from three independent experiments.
LX2343 suppressed tauopathy involving GSK-3β enzymatic activity inhibition
LX2343 restrained tau phosphorylation
Considering the association of OS with tauopathy, we examined the potential of LX2343 to suppress tau hyperphosphorylation at multiple sites, including serine 396 and 199, and threonine 23128. As expected, the results indicated that LX2343 efficiently attenuated the STZ-induced elevation of tau phosphorylation in both SH-SY5Y (Figure 5A, 5B) and primary neuronal cells (Figure 5C, 5D).

LX2343 ameliorated tau pathology involving GSK-3β inhibition. (A–D) Western blot and quantification results demonstrated that LX2343 reduced tau phosphorylation at sites of serine 396/199 and threonine 231 in SH-SY5Y and primary neuronal cells (one-way ANOVA, Dunnett's multiple comparison test, *P<0.05, **P<0.01 versus STZ. #P<0.05, ##P<0.01 versus DMSO). (E–H) Western blot and quantification results demonstrated that LX2343 reduced GSK-3β phosphorylation at tyrosine 216 and increased GSK-3β phosphorylation at serine 9 in SH-SY5Y and primary neuronal cells (one-way ANOVA, Dunnett's multiple comparison test. *P<0.05, **P<0.01 versus STZ. ##P<0.01 versus DMSO). (I) LX2343 dose-dependently inhibited GSK-3β activity in vitro (one-way ANOVA, Dunnett's multiple comparison test. *P<0.05, **P<0.01 versus DMSO; SB216763: t test. ##P<0.01 versus DMSO). SB216763: GSK-3β inhibitor. (J) LX2343 dose-dependently inhibited GSK-3β in the presence of the indicated concentrations of ATP. In the presence of 10 μmol/L of ATP, the IC50 of LX2343 was 1.37±0.47 μmol/L; in the presence of 30 μmol/L ATP, the IC50 of LX2343 was 1.84±0.07 μmol/L; and in the presence of 100 μmol/L ATP, the IC50 of LX2343 was 1.42±0.09 μmol/L. The LX2343 concentration was expressed in the log10 scale. All values were presented as the mean±SEM (n=3). GAPDH was used as the loading control in the Western blot assays. Data were obtained from three independent experiments.
LX2343 was a non-ATP competitive GSK-3β inhibitor
It is reported that phosphorylation of tau is regulated by a series of kinases and phosphatases28. Glycogen synthase kinase-3β (GSK-3β) and cyclin-dependent kinase 5 (CDK5) are two proline-directed tau kinases that phosphorylate tau at a large number of serine and threonine residues, thus playing an important role in the tau pathology of AD29,30. GSK-3β is activated through phosphorylation of Tyr 216 and inhibited by phosphorylation of Ser 929. CDK5 alone is an inactive catalytic subunit that can be activated by p35 or p25 (the cleaved product of p35)30.
Next, we detected the phosphorylation level of GSK-3β in both SH-SY5Y and primary neuronal cells using lithium chloride (LiCl) as a positive control31. The results demonstrated that both LX2343 and LiCl increased the phosphorylation at Ser 9 and decreased the phosphorylation at Tyr 216 of GSK-3β in both SH-SY5Y (Figure 5E, 5F) and primary neuronal cells (Figure 5G, 5H), indicating an alleviation of GSK-3β enzymatic activity29. Furthermore, the in vitro GSK-3β enzymatic activity assay results further verified the inhibition of the GSK-3β enzyme by LX2343 with an IC50 of 1.84±0.07 μmol/L (Figure 5I, SB216763 is a positive control32). Furthermore, to test whether competition exists between LX2343 and ATP, we investigated the effects of different concentrations of ATP on the inhibitory activity of the compound against the enzyme. The results demonstrated that inhibition of GSK-3β by LX2343 was virtually unaffected even at a high concentration of ATP (Figure 5J), which suggested that LX2343 is a non-ATP competitive inhibitor of GSK-3β. Notably, LX2343 had no effect on the protein level of either CDK5 or p35/p25 (Figure 6A–6D), and the results of the in vitro assay also suggested that LX2343 had no effect on CDK5/p25 kinase enzyme activity (Figure 6E, PHA-793887 is a positive control33).

LX2343 had no effects on CDK5 pathway. (A–D) Western blot and quantification for the protein level of CDK5, p35 and p25 in SH-SY5Y and primary neuronal cells lysate (one-way ANOVA, Dunnett's multiple comparison test, compared versus STZ, #P<0.05, ##P<0.01 versus DMSO). (E) LX2343 had no effects on CDK5/p25 activity in vitro. PHA-793887: CDK5 inhibitor (one-way ANOVA, Dunnett's multiple comparison test, P=0.8967 versus DMSO; PHA-793887, t test, **P<0.01 versus DMSO). All values were presented as mean±SEM (n=3). GAPDH was used as loading control in Western blot assays. Data were obtained from three independent experiments.
Taken together, LX2343 suppressed tauopathy by inhibiting GSK-3β enzymatic activity.
LX2343 ameliorated learning and memory impairments in ICV-STZ rats
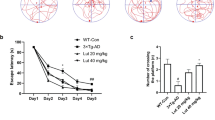
ICV injection of rats with a sub-diabetogenic dose of STV has been shown to be closely linked to the sporadic dementia of Alzheimer's disease34, which is characterized by cognitive impairment35, OS35, impaired glucose metabolism36, and hyperphosphorylated tau37. Given the pathological features of ICV-STZ rats and the efficiency of LX2343 in both protecting neuronal cells and attenuating tau pathology, we performed the MWM assay to examine the potential of LX2343 in the amelioration of memory impairment in ICV-STZ rats. To evaluate the ameliorative effects of LX2343 in AD pathologies, we chose SV as the positive control mimicking the AD pathological conditions. The results indicated that the latency time for the rats to find the hidden platform after undergoing the four-day training trials was reduced in all groups, but STZ-lesioned (SV) rats presented a longer latency time than the vehicle group (V), which suggested that the poorer learning performance was due to the ICV-STZ infusion administered to these rats (Figure 7B). Notably, LX2343 treatment improved the learning performance in the SL and SH groups when compared to the SV group. The distances swum (path lengths) by the rats while trying to find the platform on the fourth day are shown in Figure 7A. Similar to the latency time results, the path length for the rats in the SV group was longer than that for the V group, but the path length could be shortened in both the SL and SH group with LX2343 administration. According to the results of the probe trial, the rats in the SV group failed to remember the location of the platform: these rats spent less time in the target quadrant and crossed the target quadrant less frequently than the rats in the V group (Figure 7C, 7D). However, all those deficits were improved with the administration of LX2343, which suggests that LX2343 ameliorated learning and memory deficits in ICV-STZ rats.

LX2343 effectively improved memory impairment in ICV-STZ rats. (A–D) Behavioral test and quantitative analyses in ICV-STZ rats. (A) Representative tracing graphs of the training trials. (B) Escape latency during platform trials in the MWM (Two-way ANOVA with repeated measures over time: treatment, *P<0.05, **P<0.01 versus SV. ##P<0.01 versus V). (C) Representative tracing graphs of the probe trials. (D) Times of platform crossing in probe trials (one-way ANOVA, Dunnett's multiple comparison test, n=10. *P<0.05, **P<0.01 versus SV. #P<0.05 versus V). V: Rats treated with vehicle, SV: Rats injected with STZ and treated with vehicle, SL: Rats injected with STZ and treated with 7 mg·kg−1·d−1 LX2343, SH: Rats injected with STZ and treated with 21 mg·kg−1·d−1 LX2343. All values were presented as the mean±SEM (n=10 per group).
LX2343 inhibited neuronal apoptosis in ICV-STZ rats
Positive cells with apoptotic nuclei showed a green-fluorescent stain after TUNEL staining. The results demonstrated that only a few TUNEL-positive cells were present in the hippocampus and cerebral cortex of normal control rats, whereas a greater number of TUNEL-positive cells were found in STZ-treated rats (Figure 8A, 8B). Notably, administration of LX2343 attenuated neuron apoptosis in STZ-treated rats (Figure 8A, 8B).

LX2343 protected against neuronal apoptosis in ICV-STZ rats by inhibiting JNK/p38 signaling pathway. (A) Representative micrographs of TUNEL-positive cells (green) in the hippocampus and cortex of ICV-STZ rats, scale bar: 100 μm. (B) Statistical analysis of A (one-way ANOVA, Dunnett's multiple comparison test. *P<0.05, **P<0.01 versus SV. ##P<0.01 versus V). (C–F) Western blot and quantification results demonstrated that LX2343 reduced JNK, c-Jun, and p38 phosphorylation in cortical and hippocampal homogenates of ICV-STZ rats (one-way ANOVA, Dunnett's multiple comparison test: *P<0.05, **P<0.01 versus SV. #P<0.05, ##P<0.01 versus V). Rats treated with vehicle, SV: Rats ICV injected with STZ and treated with vehicle, SL: Rats injected with STZ and treated with 7 mg·kg−1·d−1 LX2343, SH: Rats injected with STZ and treated with 21 mg·kg−1·d−1 LX2343. All values were presented as the mean±SEM (n=10 per group). GAPDH was used as the loading control in Western blot assays. Data were obtained from three independent experiments.
Western blot results also demonstrated that phosphorylation levels of JNK, c-Jun, and p38 were stimulated in both the cerebral cortex (Figure 8C, 8D) and hippocampus (Figure 8E, 8F) of ICV-STZ rats compared with those of control rats. Furthermore, administration of LX2343 effectively attenuated such increases in ICV-STZ rats (Figure 8C–8F).
In addition, the in vivo regulation of the Bcl-2 superfamily and caspase-3 by LX2343 administration was also confirmed in ICV-STZ rats. The results demonstrated lower expression of Bcl-2/Bcl-xl and Bad phosphorylation and higher expression of Bax and the cleaved product of caspase-3 in the SV group compared with those in the V group in both the cerebral cortex (Figure 9A, 9B) and the hippocampus (Figure 9C, 9D) of ICV-STZ rats. LX2343 administration antagonized the shifts towards the pro-apoptotic process in both the cerebral cortex and the hippocampus of ICV-STZ rats (Figure 9A–9D). Taken together, these results indicated that LX2343 effectively inhibited neuronal apoptosis in the cerebral cortex and hippocampus of ICV-STZ rats through the inhibition of the JNK/P38 and pro-apoptotic pathways.

LX2343 effectively inhibited neuronal apoptosis involving inhibition of pro-apoptotic pathways and promotion of synapse integrity. (A–D) Western blot and quantification results demonstrated that LX2343 increased Bcl-2 or Bcl-xl protein level, increased Bad phosphorylation, decreased Bax protein level, and inhibited caspase3 cleavage in cortex or hippocampus homogenates of ICV-STZ rats (one-way ANOVA, Dunnett's multiple comparison test: *P<0.05, **P<0.01 versus SV, #P<0.05, ##P<0.01 versus V). (E–H) Western blot and quantification results demonstrated that LX2343 increased synaptophysin, PSD95 or VAMP2 protein levels in cortex or hippocampus homogenates of ICV-STZ rats (one-way ANOVA, Dunnett's multiple comparison test: *P<0.05, **P<0.01 versus SV, #P<0.05, ##P<0.01 versus V). V: Rats treated with vehicle, SV: Rats ICV injected with STZ and treated with vehicle, SL: Rats injected with STZ and treated with 7 mg·kg−1·d−1 LX2343, SH: Rats injected with STZ and treated with 21 mg·kg−1·d−1 LX2343. All values were presented as mean±SEM (n=10 per group). GAPDH was used as loading control in Western blot assays. Data were obtained from three independent experiments.
Moreover, we confirmed that the protein levels of PSD95, synaptophysin, and VAMP2 were lower in the STZ-injured rat group compared to the normal control group (Figure 9E–9H). Notably, administration of LX2343 efficiently ameliorated the STZ-induced suppression of PSD95, synaptophysin and VAMP2 protein levels in both the cerebral cortex (Figure 9E, 9F) and the hippocampus (Figure 9G, 9H) of ICV-STZ injected rats. These results support the conclusion that LX2343 alleviated synaptic plasticity damage in STZ-ICV rats.
LX2343 attenuated tau hyperphosphorylation and GSK-3β phosphorylation in ICV-STZ rats
To investigate the regulation of tau pathology by LX2343 in ICV-STZ rats, phosphorylation of tau was first evaluated by immunohistochemistry. The results (Figure 10A, 10B) noticeably indicated immuno-reactive tau phosphorylation (white arrow) in the brains of ICV-STZ rats with higher fluorescence intensity compared to that in normal rats, and the administration of LX2343 to the rats in the SL and SH groups repressed the STZ-induced increase in tau phosphorylation. Next, a Western blot assay of homogenates of the cerebral cortex (Figure 10C, 10D) and the hippocampus (Figure 10E, 10F) was also applied to evaluate the alleviation of tau phosphorylation by LX2343 in ICV-STZ rats. As expected, LX2343 administration in both the SL and SH groups apparently antagonized the STZ-induced increase in tau phosphorylation at multiple sites, including serine 396, serine 199, and threonine 231. In addition, the Western blot results of the cerebral cortical (Figure 10G, 10H) and hippocampal (Figure 10I, 10J) homogenates also indicated that LX2343 administration in both the SL and SH groups stimulated the inactive form of GSK-3β (P-S9) but suppressed the active form of GSK-3β (P-Y216) in ICV-STZ rats.

LX2343 attenuated tau pathology in ICV-STZ rats. (A) P396-tau in the brain of ICV-STZ rats was determined using immunohistochemistry, scale bar: 100 μm. (B) Statistical analysis of A (one-way ANOVA, Dunnett's multiple comparison test, **P<0.01 versus SV. ##P<0.01 versus V). (C–F) Western blot and quantification results demonstrated that LX2343 reduced phosphorylation of tau in cortical and hippocampal homogenates of ICV-STZ rats (one-way ANOVA, Dunnett's multiple comparison test: *P<0.05, **P<0.01 versus SV. #P<0.05, ##P<0.01 versus V). (G–J) Western blot and quantification demonstrated that LX2343 reduced the activity of GSK-3β in cortical and hippocampal homogenates of ICV-STZ rats (one-way ANOVA, Dunnett's multiple comparison test: *P<0.05, **P<0.01 versus SV. #P<0.05, ##P<0.01 versus V). V: Rats treated with vehicle, SV: Rats ICV injected with STZ and treated with vehicle, SL: Rats injected with STZ and treated with 7 mg·kg−1·d−1 LX2343, SH: Rats injected with STZ and treated with 21 mg·kg−1·d−1 LX2343. All values were presented as the mean±SEM (n=10 per group). GAPDH was used as the loading control in Western blot assays. Data were obtained from three independent experiments.
Discussion
AD is one of the most devastating diseases leading to an irreversible progressive cognitive impairment and is a serious threat to the elderly. However, the discovery of effective anti-AD drugs is full of challenges because of the complexity of AD pathogenesis. In our previous study10, we determined that small molecule LX2343 effectively ameliorated the cognitive deficits in APP/PS1 mice, targeting both Aβ production and clearance. Here, we also demonstrated the capability of LX2343 to treat ICV-STZ model rats through both neuronal protection and tau inhibition. Therefore, the triple effects of LX2343 on neuroprotection, Aβ inhibition, and tau modulation indicate the potential of this multi-target compound in the treatment of AD.
OS is a pivotal cellular response to the damage caused by tau toxicity, and the vicious interaction between these two causative events promotes AD progression38. Studies have identified OS as an early marker of tauopathy since OS and mitochondrial dysfunction are detected prior to tau hyperphosphorylation and NFT accumulation in tauP301S transgenic mice, which show tau hyperphosphorylation and tangle formation in 3 months39. On the one hand, OS amplifies tau-induced neurotoxicity by induction of apoptosis and dysregulation of cell energy metabolism and promotes tau hyperphosphorylation and aggregation38. On the other hand, tauopathy induces OS40. Some OS markers are notably increased in tauopathy, such as malondialdehyde and 4-hydroxynonenal41,42,43. The activation of antioxidant defenses is also observed in tauopathy44,45. Such a positive feed-forward loop of OS and tauopathy greatly promotes AD progression and makes its pathogenesis more intricate.
In view of the close link between OS and tauopathy, many antioxidants have recently been discovered that protect against tauopathy in different AD models. For example, vitamin E decreased ROS production and tau phosphorylation22,46 and chronic administration of coenzyme Q10 to tauP301S mice alleviated ROS accumulation and tau hyperphosphorylation, thus improving behavioral deficits in the transgenic mice40. Although the results obtained using animal studies have suggested that antioxidants are potential therapeutic agents for the treatment of AD and tauopathy-relevant diseases, the translation of these inspiring results obtained in animal models into clinical use has not yet led to significant advances38. Therefore, new strategies are needed to design effective agents against OS or tauopathy to break the deadlock. Here, we discovered that LX2343 was able to ameliorate both OS and tauopathy at the same time; LX2343 substantially reduced ROS accumulation similar to an antioxidant and restored mitochondrial function through stabilizing the mitochondrial membrane potential, maintaining mitochondrial morphological integrity, and increasing ATP biosynthesis. In addition, LX2343 also suppressed hyperphosphorylation of tau by inhibition of GSK-3β. Thus, the dual effect of LX2343 has emphasized its potential for breaking the cycle of OS and tauopathy.
GSK-3β as an isoform of GSK-3 can phosphorylate tau at several sites, and its dysregulation is responsible for tau hyperphosphorylation. In addition to the regulation of tau, GSK-3β also plays a pivotal role in other cellular functions47,48. It is reported that GSK-3β regulates apoptotic pathways directly or indirectly. It directly triggers the mitochondrial apoptosis cascade by promoting Bax translocation to mitochondria49 and indirectly leads to canonical Wnt pathway-mediated apoptosis through phosphorylation of β-catenin50. Here, we also investigated the regulatory effects of LX2343-mediated GSK-3β inhibition against apoptosis. Interestingly, Western blot results demonstrated a decrease in Bax translocation to mitochondria with LX2343 treatment (Supplementary Figure S3A), but no effects on the Wnt pathway were determined as LX2343 did not alleviate the phosphorylation of β-catenin (Supplementary Figure S3B). These results indicated that the direct inhibition of GSK-3β by LX2343 might contribute, at least partly, to the efficacy of LX2343 in neuroprotection.
Collectively, our data showed that small molecule LX2343 effectively improved cognitive dysfunction in ICV-STZ AD model rats by suppression of both OS-induced neuronal apoptosis and tauopathy. The underlying mechanisms have been intensively investigated. As shown in Figure 11, LX2343 rescued neuronal cells from apoptosis by maintaining the integrity of mitochondrial function and morphology, alleviating OS and the JNK/p38 pathway and regulating anti- and pro-apoptotic proteins. In addition, LX2343 also potently inhibited tau hyperphosphorylation by functioning as a non-ATP competitive inhibitor of GSK-3β. Combined with our previous finding10, our current work has largely enriched our knowledge of the pharmacological efficacy of LX2343 in the treatment of AD.

A proposed model illustrating the mechanism underlying the efficiency of LX2343 in the amelioration of cognitive impairment in AD. LX2343 rescued neuronal cells from apoptosis by maintaining the integrity of mitochondrial function and morphology, alleviating the OS and JNK/p38 pathway and regulating anti- and pro-apoptotic proteins. In addition, LX2343 also inhibited tau hyperphosphorylation by functioning as a non-ATP competitive inhibitor of GSK-3β.
Author contribution
Xu SHEN, Zhi-yuan ZHU, and Xiao-dan GUO designed the experiment; Xiao-dan GUO carried out the experiments and analyzed the data; Guang-long SUN, Vatcharin RUKACHAISIRIKUL, and Li-hong HU synthesized the compound; Ting-ting ZHUO, Xin XU, Xiao-fan SHI, and Yi-yang Wang participated in animal sacrifice; Xu SHEN and Xiao-dan GUO wrote the manuscript.
References
Hardy J . A hundred years of Alzheimer's disease research. Neuron 2006; 52: 3–13.
Sosa-Ortiz AL, Acosta-Castillo I, Prince MJ . Epidemiology of dementias and Alzheimer's disease. Arch Med Res 2012; 43: 600–8.
Walsh DM, Selkoe DJ . Deciphering the molecular basis of memory failure in Alzheimer's disease. Neuron 2004; 44: 181–93.
Misra S, Medhi B . Drug development status for Alzheimer's disease: present scenario. Neurol Sci 2013; 34: 831–9.
Clark TA, Lee HP, Rolston RK, Zhu X, Marlatt MW, Castellani RJ, et al. Oxidative stress and its implications for future treatments and management of Alzheimer disease. Int J Biomed Sci 2010; 6: 225–7.
Guan ZZ . Cross-talk between oxidative stress and modifications of cholinergic and glutaminergic receptors in the pathogenesis of Alzheimer's disease. Acta Pharmacol Sin 2008; 29: 773–80.
Naini SM, Soussi-Yanicostas N . Tau hyperphosphorylation and oxidative stress, a critical vicious circle in neurodegenerative tauopathies? Oxid Med Cell Longev 2015; 2015: 151979.
Kempf M, Clement A, Faissner A, Lee G, Brandt R . Tau binds to the distal axon early in development of polarity in a microtubule- and microfilament-dependent manner. J Neurosci 1996; 16: 5583–92.
Alonso A, Zaidi T, Novak M, Grundke–Iqbal I, Iqbal K . Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments. Proc Natl Acad Sci U S A 2001; 98: 6923–8.
Guo XD, Sun GL, Zhou TT, Xu X, Zhu ZY, Rukachaisirikul V, et al. Small molecule LX2343 ameliorates cognitive deficits in AD model mice by targeting both amyloid beta production and clearance. Acta Pharmacol Sin 2016; 37: 1281–97.
Salkovic-Petrisic M, Knezovic A, Hoyer S, Riederer P . What have we learned from the streptozotocin-induced animal model of sporadic Alzheimer's disease, about the therapeutic strategies in Alzheimer's research. J Neural Transm (Vienna) 2013; 120: 233–52.
Zhu Z, Yan J, Jiang W, Yao XG, Chen J, Chen L, et al. Arctigenin effectively ameliorates memory impairment in Alzheimer's disease model mice targeting both beta-amyloid production and clearance. J Neurosci 2013; 33: 13138–49.
Zhong H, Zou H, Semenov MV, Moshinsky D, He X, Huang H, et al. Characterization and development of novel small-molecules inhibiting GSK3 and activating Wnt signaling. Mol Biosyst 2009; 5: 1356–60.
Mehla J, Pahuja M, Gupta YK . Streptozotocin-induced sporadic Alzheimer's disease: selection of appropriate dose. J Alzheimers Dis 2013; 33: 17–21.
Zhu Z, Li C, Wang X, Yang Z, Chen J, Hu L, et al. 2,2′,4′-trihydroxychalcone from Glycyrrhiza glabra as a new specific BACE1 inhibitor efficiently ameliorates memory impairment in mice. J Neurochem 2010; 114: 374–85.
Gold R, Schmied M, Giegerich G, Breitschopf H, Hartung HP, Toyka KV, et al. Differentiation between cellular apoptosis and necrosis by the combined use of in situ tailing and nick translation techniques. Lab Invest 1994; 71: 219–25.
Biswas J, Goswami P, Gupta S, Joshi N, Nath C, Singh S . Streptozotocin induced neurotoxicity involves Alzheimer's related pathological markers: a study on N2A cells. Mol Neurobiol 2016; 53: 2794–806.
Rai S, Kamat PK, Nath C, Shukla R . Glial activation and post-synaptic neurotoxicity: the key events in Streptozotocin (ICV) induced memory impairment in rats. Pharmacol Biochem Behav 2014; 117: 104–17.
Valtorta F, Pennuto M, Bonanomi D, Benfenati F . Synaptophysin: leading actor or walk-on role in synaptic vesicle exocytosis? BioEssays 2004; 26: 445–53.
Reddy PH, Reddy TP . Mitochondria as a therapeutic target for aging and neurodegenerative diseases. Curr Alzheimer Res 2011; 8: 393–409.
Ortiz Mdel C, Lores-Arnaiz S, Albertoni Borghese MF, Balonga S, Lavagna A, Filipuzzi AL, et al. Mitochondrial dysfunction in brain cortex mitochondria of STZ-diabetic rats: effect of l-Arginine. Neurochem Res 2013; 38: 2570–80.
Nakashima H, Ishihara T, Yokota O, Terada S, Trojanowski JQ, Lee VM, et al. Effects of alpha–tocopherol on an animal model of tauopathies. Free Radic Biol Med 2004; 37: 176–86.
Leuner K, Schulz K, Schutt T, Pantel J, Prvulovic D, Rhein V, et al. Peripheral mitochondrial dysfunction in Alzheimer's disease: focus on lymphocytes. Mol Neurobiol 2012; 46: 194–204.
Son Y, Cheong YK, Kim NH, Chung HT, Kang DG, Pae HO . Mitogen–activated protein kinases and reactive oxygen species: How can ROS activate MAPK pathways? J Signal Transduct 2011; 2011: 792639.
Chen Z, Zhong C . Decoding Alzheimer's disease from perturbed cerebral glucose metabolism: implications for diagnostic and therapeutic strategies. Prog Neurobiol 2013; 108: 21–43.
Hanson CJ, Bootman MD, Distelhorst CW, Maraldi T, Roderick HL . The cellular concentration of Bcl-2 determines its pro- or anti-apoptotic effect. Cell Calcium 2008; 44: 243–58.
Taylor RC, Cullen SP, Martin SJ . Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol 2008; 9: 231–41.
Ishiguro K, Sato K, Takamatsu M, Park J, Uchida T, Imahori K . Analysis of phosphorylation of tau with antibodies specific for phosphorylation sites. Neurosci Lett 1995; 202: 81–4.
Wang JZ, Wu Q, Smith A, Grundke-Iqbal I, Iqbal K . Tau is phosphorylated by GSK-3 at several sites found in Alzheimer disease and its biological activity markedly inhibited only after it is prephosphorylated by A-kinase. FEBS Lett 1998; 436: 28–34.
Kimura T, Ishiguro K, Hisanaga S . Physiological and pathological phosphorylation of tau by Cdk5. Front Mol Neurosci 2014; 7: 65.
Aghdam SY, Barger SW . Glycogen synthase kinase-3 in neurodegeneration and neuroprotection: lessons from lithium. Curr Alzheimer Res 2007; 4: 21–31.
Gao L, Zhao M, Li P, Kong J, Liu Z, Chen Y, et al. Glycogen synthase kinase 3 (GSK3)-inhibitor SB216763 promotes the conversion of human umbilical cord mesenchymal stem cells into neural precursors in adherent culture. Hum Cell 2017; 30: 11–22.
Nguyen TK, Grant S . Dinaciclib (SCH727965) inhibits the unfolded protein response through a CDK1- and 5-dependent mechanism. Mol Cancer Ther 2014; 13: 662–74.
Agrawal R, Tyagi E, Shukla R, Nath C . A study of brain insulin receptors, AChE activity and oxidative stress in rat model of ICV STZ induced dementia. Neuropharmacology 2009; 56: 779–87.
Sharma M, Gupta YK . Intracerebroventricular injection of streptozotocin in rats produces both oxidative stress in the brain and cognitive impairment. Life Sci 2001; 68: 1021–9.
Lannert H, Wirtz P, Schuhmann V, Galmbacher R . Effects of Estradiol (-17beta) on learning, memory and cerebral energy metabolism in male rats after intracerebroventricular administration of streptozotocin. J Neural Transm (Vienna) 1998; 105: 1045–63.
Lee CW, Shih YH, Wu SY, Yang T, Lin C, Kuo YM . Hypoglycemia induces tau hyperphosphorylation. Curr Alzheimer Res 2013; 10: 298–308.
Alavi Naini SM, Soussi-Yanicostas N . Tau hyperphosphorylation and oxidative stress, a critical vicious circle in neurodegenerative tauopathies? Oxid Med Cell Longev 2015; 2015: 151979.
Dumont M, Stack C, Elipenahli C, Jainuddin S, Gerges M, Starkova NN, et al. Behavioral deficit, oxidative stress, and mitochondrial dysfunction precede tau pathology in P301S transgenic mice. FASEB J 2011; 25: 4063–72.
Elipenahli C, Stack C, Jainuddin S, Gerges M, Yang L, Starkov A, et al. Behavioral improvement after chronic administration of coenzyme Q10 in P301S transgenic mice. J Alzheimers Dis 2012; 28: 173–82.
Martinez A, Carmona M, Portero-Otin M, Naudi A, Pamplona R, Ferrer I . Type-dependent oxidative damage in frontotemporal lobar degeneration: cortical astrocytes are targets of oxidative damage. J Neuropathol Exp Neurol 2008; 67: 1122–36.
Albers DS, Augood SJ, Martin DM, Standaert DG, Vonsattel JP, Beal MF . Evidence for oxidative stress in the subthalamic nucleus in progressive supranuclear palsy. J Neurochem 1999; 73: 881–4.
Odetti P, Garibaldi S, Norese R, Angelini G, Marinelli L, Valentini S, et al. Lipoperoxidation is selectively involved in progressive supranuclear palsy. J Neuropathol Exp Neurol 2000; 59: 393–7.
Cantuti-Castelvetri I, Keller-McGandy CE, Albers DS, Beal MF, Vonsattel JP, Standaert DG, et al. Expression and activity of antioxidants in the brain in progressive supranuclear palsy. Brain Res 2002; 930: 170–81.
Aoyama K, Matsubara K, Kobayashi S . Aging and oxidative stress in progressive supranuclear palsy. Eur J Neurol 2006; 13: 89–92.
Cente M, Filipcik P, Mandakova S, Zilka N, Krajciova G, Novak M . Expression of a truncated human tau protein induces aqueous-phase free radicals in a rat model of tauopathy: implications for targeted antioxidative therapy. J Alzheimers Dis 2009; 17: 913–20.
Embi N, Rylatt DB, Cohen P . Glycogen synthase kinase-3 from rabbit skeletal muscle. Separation from cyclic-AMP-dependent protein kinase and phosphorylase kinase. Eur J Biochem 1980; 107: 519–27.
Forde JE, Dale TC . Glycogen synthase kinase 3: a key regulator of cellular fate. Cell Mol Life Sci 2007; 64: 1930–44.
Linseman DA, Butts BD, Precht TA, Phelps RA, Le SS, Laessig TA, et al. Glycogen synthase kinase-3beta phosphorylates Bax and promotes its mitochondrial localization during neuronal apoptosis. J Neurosci 2004; 24: 9993–10002.
Jacobs KM, Bhave SR, Ferraro DJ, Jaboin JJ, Hallahan DE, Thotala D . GSK-3beta: a bifunctional role in cell death pathways. Int J Cell Biol 2012; 2012: 930710.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81220108025 and 81473141), NSFC-TRF collaboration projects (81561148011 and DBG5980001), the Drug Innovation Project of SIMM (CASIMM0120154035) and Personalized Medicines-Molecular Signature-based Drug Discovery and Development, Strategic Priority Research Program of the Chinese Academy of Sciences (XDA12040303).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Supplementary information is available on the website of Acta Pharmacologica Sinica.
Supplementary information
Supplementary Figure S1
No significant differences between normal control group and shame group in cognitive function. (JPG 394 kb)
Supplementary Figure S2
Combination of LX2343 with VE exhibited a synergistic action in neuroprotection. (JPG 635 kb)
Supplementary Figure S3
LX2343 inhibited p53 translocation to mitochondria, but had no effects on β-catenin. (JPG 755 kb)
Supplementary Table S1
The exact values of mean±SEM. (DOC 228 kb)
PowerPoint slides
Rights and permissions
About this article

Cite this article
Guo, Xd., Sun, Gl., Zhou, Tt. et al. LX2343 alleviates cognitive impairments in AD model rats by inhibiting oxidative stress-induced neuronal apoptosis and tauopathy. Acta Pharmacol Sin 38, 1104–1119 (2017). https://doi.org/10.1038/aps.2016.128
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2016.128

{kind=link}
{kind=link}
{kind=link}