
Abstract
Oblongifolin C (OC) and guttiferone K (GUTK) are two anticancer compounds extracted from Garcinia yunnanensis Hu, but they act by different mechanisms. In this study we investigated whether a combination of OC and GUTK (1:1 molar ratio) could produce synergistic anticancer effects against human colorectal cancer cells in vitro. For comparison, we also examined the anticancer efficacy of ethanol extracts from G yunnanensis fruit, which contain OC and GUTK up to 5%. Compared to OC and GUTK alone, the combination of OC and GUTK as well as the ethanol extracts more potently inhibited the cancer cell growth with IC50 values of 3.4 μmol/L and 3.85 μg/mL, respectively. Furthermore, OC and GUTK displayed synergistic inhibition on HCT116 cells: co-treatment with OC and GUTK induced more prominent apoptosis than treatment with either drug alone. Moreover, the combination of OC and GUTK markedly increased cleavage of casapse-3 and PARP, and enhanced cellular ROS production and increased JNK protein phosphorylation. In addition, the combination of OC and GUTK exerted stronger effects under nutrient-deprived conditions than in complete medium, suggesting that autophagy played an essential role in regulating OC- and GUTK-mediated cell death. OC and GUTK are the main components that contribute to the anticancer activity of G yunnanensis and the compounds have apoptosis-inducing effects in HCT116 cells in vitro.
Similar content being viewed by others

Introduction
Colorectal cancer is the third most common cancer in men and the second most common cancer in women worldwide. In 2012, approximately 1.4 million cases of colorectal cancer were diagnosed, and approximately 694 000 cancer-related deaths were due to colorectal cancer. The prognosis of this disease is poor, and the mortality is as high as 52% in less developed regions. Currently, the treatment options for patients with colorectal cancer are surgery and chemotherapy1,2.
Macroautophagy (hereafter referred to as “autophagy”) and apoptosis are two types of cell death that play important roles in cancer therapy3,4, and reactive oxygen species (ROS) may stimulate cell death via autophagy and apoptosis. ROS-inducing reagents that elevate oxidative stress often act as potential anticancer reagents5,6. Specifically, C-Jun N-terminal kinase (JNK) signaling is involved in the response to oxidative stress, which plays a crucial role in the induction of autophagy and apoptosis7,8.
Compounds and extracts from natural products are important sources of chemopreventive and therapeutic agents in cancer treatment9,10,11. For example, natural polyisoprenylated benzophenones (PPBS) possess a wide range of biological activities, including cytotoxic, anti-viral, anti-oxidant, and anti-inflammatory properties, and the members of the Guttiferae family of plants are characterized by their ability to produce large amounts of benzophenones12,13. Specifically, oblongifolin C (OC) and guttiferone K (GUTK) are the main active components of Garcinia yunnanensis Hu14, and our previous studies have shown that OC has multiple anticancer activities, including the promotion of apoptosis and inhibition of autophagy and metastasis15,16,17. Similarly, we found that GUTK activates apoptosis, arrests the cell cycle, and promotes autophagy18,19. Because OC and GUTK target different cell death pathways and are the main active compounds in Garcinia yunnanensis Hu, we sought to determine whether the combination of OC and GUTK might synergistically kill cancer cells. Furthermore, we also investigated the ability of OC- and GUTK-containing crude extracts to efficiently induce cell death. Our results showed that the combination of OC and GUTK has therapeutic potential as an anticancer treatment. Interestingly, crude extracts containing approximately equal amounts of OC and GUTK also strongly inhibited HCT116 cell growth, thus suggesting that this extract has the potential to be developed as a low-cost anticancer drug from plants.
Materials and methods
Cell line
HCT116 human colon cancer cells were purchased from the American Type Culture Collection. The cells were cultured in DMEM (Gibco/Invitrogen, 12800-017) supplemented with 10% fetal bovine serum (Invitrogen) and 1% penicillin/streptomycin (Gibco/Invitrogen, 15140-122) and were maintained at 37 °C in a humidified incubator under 5% CO2.
Preparation of G yunnanensis extract and fractions
The fruits of Garcinia yunnanensis were collected from the Yunnan province in China. The plant material was authenticated by Prof Hong WANG of the Xishuangbanna Tropical Botanical Garden at the Chinese Academy of Sciences. A voucher specimen was deposited in the herbarium of the Innovative Research Laboratory of TCM at the Shanghai University of Traditional Chinese Medicine. The fruits of G yunnanensis were ground into a fine powder. A 100.0×g sample of fine powder was soaked in 500 mL of 95% ethanol (EtOH, Sinopharm Chemical Reagent Co, Ltd, Cat 10009171) for 24 h at room temperature, extracted with 95% EtOH (3×500 mL) at 90 °C in a water bath for 1 h, and filtered. The combined filtrate was evaporated to a volume of 350 mL under a vacuum. The extract was passed through a D101 resin column gradient and eluted with EtOH, and the 0%–70% EtOH eluent was concentrated to produce fraction A. The 70%–95% EtOH eluent was concentrated to produce fraction B, which contained the majority of oblongifolin C and guttiferone K. The ethanolic G yunnanensis fruit extract fraction A (GYFA, NO OC+GUTK fraction) and fraction B (GYFB, OC+GUTK fraction) were dissolved in DMSO (Sigma, Cat. D2650) prior to each assay. Oblongifolin C and guttiferone K were isolated from G. yunnanensis in our laboratory and were identified on the basis of mass spectrometry and nuclear magnetic resonance spectroscopy.
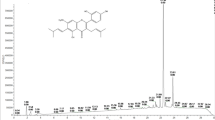
GYFB was analyzed by HPLC with a Waters e2695 (Waters, America) separation module and an Agilent ZORBAX column (SB-C18, 5 μm, 4.6×250 mm). The mobile phase consisted of (A) acetonitrile (Sinopharm Chemical Reagent Co, Ltd, Cat 40064193) and (B) 0.1% formic acid in water. The HPLC elution conditions were as follows: 80%–95% A (0-10 min), 95%–100% A (10–15 min), 100%–95% A (15–20 min), and 95%–80% A (20–35 min). The flow rate was 1.0 mL/min. The column was maintained at 35 °C, and the detection wavelength was set to 349 nm.
MTT assay
Cells were seeded in a 96-well plate at a density of 4×103 cells per 100 μL per well. After overnight incubation, the cells were treated with different concentrations of the indicated compounds for 48 h, and 10 μL of 5 mg/mL 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solution was then added to each well and incubated for 4 h at 37 °C. Subsequently, the culture medium was removed, and 100 μL of DMSO was added to each well. The absorbance was measured at 570 nm, and the cell viability was normalized and is presented as a percentage of the control. The experiments were performed in triplicate, and the absorbance for each treatment condition is presented as a percentage of the control condition.
Combined effect evaluation
The interaction effect between OC and GUTK was analyzed using the combination index (CI)20,21. The CI value is defined by the following equation: CI=(D)1/(Dx)1+(D)2/(Dx)2, where (Dx)1 and (Dx)2 are the concentrations for D1 (OC) and D2 (GUTK) alone that inhibit x% cell growth, and (D)1 and (D)2 are the concentrations of OC and GUTK in combination that result in identical cell growth inhibition. The CI value was divided into three categories, CI<1, CI=1, and CI > 1, indicating synergistic, additive, and antagonistic effects, respectively. Each CI value is the mean value of three independent experiments. The data were analyzed using Calcusyn software.
Flow cytometry
Cells were seeded in 6-well plates and incubated overnight before the addition of various concentrations of compounds in complete (DMEM with serum) or nutrient-deprived (EBSS) medium for the indicated times. For the cell cycle distribution analysis, cells were washed with PBS and fixed in 70% ice-cold ethanol overnight. They were then suspended in PI (50 μg/mL) and RNase A (50 μg/mL), and the DNA content of samples was subsequently analyzed by flow cytometry.
Apoptosis was analyzed with an Annexin V-FITC/PI Cell Apoptosis Detection kit. Briefly, cells were suspended in binding buffer and stained with Annexin V-FITC/PI for 15 min in the dark. The stained cells were quantified by flow cytometry, which was conducted on a BD Influx™ instrument (BD Biosciences). All flow cytometry data were analyzed with FlowJo 7.6.1 software18.
Western blotting
The cells were treated at the indicated concentrations, and both floating and adherent cells were harvested before cells were lysed with radio-immunoprecipitation (RIPA) buffer containing protease and phosphatase inhibitors. The protein concentrations were determined using a BCA protein assay, and equivalent amounts of total protein were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS/PAGE) and transferred to polyvinylidenedifluoride (PVDF, Millipore Corp, Billerica, MA, USA) membranes. PVDF membranes were washed and blocked for 1 h with 5% (w/v) dry milk in TBST at room temperature with shaking. The membranes were blotted overnight with primary antibodies in 5% BSA in TBST at 4°C with shaking. The primary antibodies to the following proteins were used: PARP, caspase-3, phospho-SAPK/JNK, α-Tubulin, LC3B, and SQSTM1/p62 (Cell Signaling Technology, Inc, Santa Cruz Biotechnology Inc, Sigma-Aldrich, and MBL). The membranes were washed and probed with horseradish peroxidase-conjugated secondary anti-mouse or anti-rabbit antibodies diluted in 5% nonfat dry milk in TBST for 1 h. Protein bands were detected using an ImageQuant LAS 4000 Mini instrument (GE Healthcare).
Clonogenic assay
HCT116 cells were plated at 500 cells per well in 6-well plates. After 24 h, the cells were treated with the compounds at the indicated concentrations for 24, 48, or 72 h. The cells were then washed with PBS and fed with fresh culture medium before being allowed to form colonies for 14 days. Subsequently, the cells were washed three times with PBS, fixed with ethanol, and stained with 0.04% crystal violet. Colonies of at least 50 cells were counted using a microscope, and the inhibition of colony formation was calculated as a percentage of the vehicle control value.
Measurement of ROS
Cells were treated with compounds at the indicated concentrations in a 6-well plate for 24 h. At the end of treatment, the cells were washed with PBS and incubated with 20 μmol/L DCFH-DA for 30 min at 37 °C in the dark. Stained cells were washed with PBS and observed under a fluorescence microscope.
Statistical analysis
All results are presented as the mean±SEM of three independent experiments. The data were statistically analyzed using a one-way ANOVA for multiple comparisons. For all statistical tests, P<0.05 was considered to indicate a significant difference.
Results
OC and GUTK are the major components of the ethanolic extract of Garcinia yunnanensis Hu that induce cell death
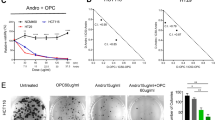
To investigate the components of the ethanolic G yunnanensis extract, we separated the extract into several fractions by using a D101 resin column with an ethanol elution gradient. The ethanol extract was clearly divided into two fractions by HPLC analysis: fraction GYFA and fraction GYFB. As shown in the flow chart, fractions GYFA (NO OC+GUTK fraction) and GYFB (OC+GUTK fraction) were easily separated with a D101 resin column (Figure 1A). Figure 1B shows a representative chromatogram of the major constituents of G yunnanensis extract. GUTK and OC were the main components of G yunnanensis extract, and this extract contained approximately equimolar amounts of these components. Among the fractions, GYFB (OC+GUTK fraction) potently inhibited HCT116 cells in a dose-dependent manner, with an IC50 value of 3.85 μg/mL, whereas GYFA (NO OC+GUTK fraction) exerted almost no effect on cell viability at a concentration of 100 μg/mL (Figure 1C), and similar results were observed by using a SYBR green assay (Supplementary Figure S1). To investigate the effect of G yunnanensis extract on cancer cell viability, we performed flow cytometry to quantify the percentage of cells in the apoptotic sub-G1 phase. GYFB (OC+GUTK fraction) treatment increased the percentage of cells in the sub-G1 phase in a dose-dependent manner, both in complete medium and in EBSS medium. GYFB (OC+GUTK fraction) treatment significantly increased the percentage of cells in the sub-G1 phase, which ranged from 2% to approximately 30%, whereas GYFA (NO OC+GUTK fraction) did not increase this percentage in either complete medium or EBSS medium (Figure 1D, 1E).

OC and GUTK are the major components of the ethanolic extract of Garcinia yunnanensis Hu, which can induce cell death. (A) The extraction flow chart of GYFA (NO OC+GUTK fraction) and GYFB (OC+GUTK fraction). (B) The UV chromatogram of GYFB (detection wavelength: 349 nm). (C) The effect of G yunnanensis extracts on colon cancer cell viability was assessed by MTT assays. HCT116 cells were treated with GYFA (0–100 μg/mL) or GYFB (0–20 μg/mL) for 48 h. The data are presented as the mean±SEM and were analyzed with a one-way ANOVA, **P<0.01 vs control.

1E. OC and GUTK are the major components of the ethanolic extract of Garcinia yunnanensis Hu, which can induce cell death. (D, E) G yunnanensis extracts induced cell death in HCT116 cells. After treatment, the sub-G1 peak was analyzed by flow cytometry with PI staining. The data are presented as the mean±SEM and were analyzed with a one-way ANOVA, *P<0.05, **P<0.01. (D) HCT-116 cells were treated with GYFA (10, 50, and 100 μg/mL) or GYFB (1, 5, and 10 μg/mL) for 48 h in complete medium. (E) HCT-116 cells were treated with GYFA (10, 50, and 100 μg/mL) or GYFB (1, 5, and 10 μg/mL) for 6 h in EBSS.
Ethanolic extract GYFB induces apoptosis by activating JNK
To further study the mechanisms by which GFYA and GFYB induce cell death, the cleavage of pro-caspase-3 and PARP as well as the expression levels of LC3-II and p62 were investigated in both complete and EBSS media. In complete medium, GYFB (10.0 μg/mL) induced the cleavage of pro-caspase-3 into its two active fragments (17 and 19 kDa), stimulated the cleavage of PARP, increased the LC3-II protein levels, decreased the p62 protein levels, and increased the expression of phosphorylated JNK without changing the amount of total JNK (Figure 2A). Similarly, in EBSS medium, GYFB (5.0 μg/mL) induced the cleavage of pro-caspase-3, stimulated the cleavage of PARP, increased the LC3-II protein levels, decreased the p62 protein levels, and increased the expression of phosphorylated JNK (Figure 2B). In contrast, GYFA treatment did not significantly change the expression or cleavage state of any examined protein (Figure 2A and 2B).

GYFB but not GYFA activated apoptosis-related proteins and regulated the phosphorylation of JNK and the levels of LC3-II and p62 in a dose-dependent manner. Tubulin was used as a loading control. (A) HCT116 cells were treated with GYFA (12.5, 25, 50, and 100 μg/mL) or (GYFB 1.25, 2.5, 5, and 10 μg/mL) for 48 h in complete medium. (B) HCT116 cells were treated with GYFA (12.5, 25, 50, and 100 μg/mL) or GYFB (1.25, 2.5, 5, and 10 μg/mL) for 6 h in EBSS.
The combination of OC and GUTK inhibits cell proliferation and induces cell death
Our data show that GYFB (OC+GUTK fraction) is primarily responsible for the anticancer activity of G yunnanensis. Both OC and GUTK (Figure 3A) are the main components of the EtOH fraction extracted from G yunnanensis, and they compose up to 5% of the EtOH fraction. Therefore, we sought to determine whether co-treatment with OC and GUTK would exhibit similar bioactivity. Indeed, OC, GUTK and their combination (1:1 molar ratio) significantly decreased the viability of HCT116 cells in a dose-dependent manner according to MTT (Figure 3B), SYBR green (Supplementary Figure S1) and LDH assays (Supplementary Figure S2). Specifically, the IC50 values of OC, GUTK and their combination were 7.5, 7.5, and 3.4 μmol/L, respectively, after 48 h of treatment. Moreover, OC and GUTK showed synergistic inhibitory effects. The CI values when OC and GUTK were co-administered at concentrations of 1.25, 1.75, 5, 7, and 14 μmol/L were 0.71, 0.79, 0.96, 0.94 and 1.00, respectively (Figure 3C). To study the potential effects of OC, GUTK and their combination on long-term proliferation, we used colony formation assays. HCT116 cells were seeded at 500 cells per well and treated with 7.5 μmol/L OC, 7.5 μmol/L GUTK, or their combination for 24, 48, or 72 h in complete medium. After 24 h, neither compound alone nor the combined treatment significantly affected colony formation, whereas the combined treatment suppressed the formation of colonies compared with treatment with OC alone or GUTK alone after 48 and 72 h of treatment (Figure 3D). To investigate whether treatment with the combination of OC and GUTK might increase the cancer cell cytotoxicity of these compounds, we performed flow cytometry to quantify apoptotic cells in the sub-G1 phase. Treatment with either OC or GUTK alone (7.5 μmol/L) in complete medium induced less than 20% of the cells to enter the sub-G1 phase, whereas combined treatment with OC and GUTK (7.5 μmol/L) increased the percentage of sub-G1 cells to 45% (Figure 3E). Moreover, treatment with either OC or GUTK alone (2 μmol/L) in EBSS medium induced less than 15% of cells to enter the sub-G1 phase, whereas combined treatment with OC and GUTK (2 μmol/L) significantly increased the sub-G1 cell population to 37% (Figure 3F). These results indicate that combined treatment with OC and GUTK is potently cytotoxic and inhibits cancer cell proliferation.

OC and GUTK in combination inhibit cell proliferation and induce cell death in HCT116 cells. (A) The chemical structures of OC and GUTK. (B) The viability of cells treated with OC combined with GUTK was detected with MTT assays. HCT116 cells were treated with OC or GUTK alone (0–15 μmol/L) or OC combined with GUTK (0–15 μmol/L) for 48 h. Values are expressed as the percentage of inhibition compared with control cultures. n=3. Data are presented as the mean±SEM and were analyzed with a one-way ANOVA. *P<0.05, **P<0.01 vs 0 μmol/L group. ##P<0.01 vs OC or GUTK alone group. (C) CI value of OC and GUTK (CI<1 indicates a synergistic effect). (D) Clonogenic assay. HCT116 cells were seeded in 6-well plates, 500 cells per well. After 24 h, the cells were treated with 7.5 μmol/L OC alone, 7.5 μmol/L GUTK alone or both compounds in combination. After 24, 48 and 72 h, cells were transferred into complete medium without drugs and cultured for another 14 d. Mean±SEM. **P<0.01 vs0 μmol/L group. ##P<0.01 vs OC or GUTK alone group. The data were analyzed with a one-way ANOVA.

OC and GUTK in combination inhibit cell proliferation and induce cell death in HCT116 cells. (E, F) OC combined with GUTK induced cell death in HCT116 cells. After treatment, the sub-G1 peak was analyzed by flow cytometry with PI staining. Data are presented as the mean±SEM and were analyzed with a one-way ANOVA. **P<0.01. (E) HCT116 cells were treated with OC (7.5 μmol/L), GUTK (7.5 μmol/L), or OC+GUTK (7.5 μmol/L each) for 48 h in complete medium. (F) HCT-116 cells were treated with OC (2 μmol/L), GUTK (2 μmol/L) or OC+GUTK (2 μmol/L each) for 3 h in EBSS.
The combination of OC and GUTK induces cell death by inducing apoptosis and autophagy
To further study the apoptotic mechanisms, the effects of OC alone, GUTK alone, and both compounds in combination on the cleavage of pro-caspase-3 and PARP, as well as the expression levels of LC3-II and p62, were investigated in both complete and EBSS medium. In complete medium, co-treatment with 7.5 μmol/L OC and GUTK induced the cleavage of pro-caspase-3 into its two active fragments (17 and 19 kDa). OC and GUTK in combination also stimulated the cleavage of PARP. Furthermore, OC and GUTK in combination increased the LC3-II protein levels and decreased the p62 protein levels (Figure 4A). In EBSS medium, the combination of OC and GUTK dramatically increased apoptosis of cancer cells. Moreover, low concentrations of the drug combination efficiently promoted caspase-3 activation and PARP cleavage and caused an increase in LC3-II protein and a decreases in p62 protein under EBSS conditions (Figure 4B and 4C). These data indicated that the combination of OC and GUTK induces apoptosis by activating caspase signaling pathways.

OC and GUTK in combination induce cell death via apoptosis and autophagy in HCT116 cells. Combined treatment with OC and GUTK activates apoptosis-related proteins and regulates the levels of LC3-II and p62. Tubulin was used as a loading control. (A) HCT116 cells were treated with OC or GUTK alone (1, 2, 5 and 7.5 μmol/L) or OC combined with GUTK (1, 2, 5 and 7.5 μmol/L) for 48 h in complete medium. (B) HCT116 cells were treated with OC or GUTK alone (1, 2 and 5 μmol/L) or OC combined with GUTK (1, 2 and 5 μmol/L) for 3 h in EBSS. (C) HCT116 cells were treated with OC alone (2 μmol/L), GUTK alone (2 μmol/L), or OC+GUTK (2 μmol/L each) for 3, 6, or 9 h in EBSS.
The autophagy inhibitor HCQ decreased the degree of apoptosis caused by the combination of OC and GUTK. As shown in Figure 5A, HCQ co-treatment caused little cell death in complete medium, whereas HCQ co-treatment rescued cell death in EBSS medium. Moreover, pre-treatment with the caspase inhibitor Z-VAD-fmk also prevented cell death in both complete medium and EBSS medium (Figure 5B). Overall, our data suggested that the combination of OC and GUTK activates apoptosis and autophagy, thus contributing to the death of HCT116 cells.

Cell death induced by OC and GUTK co-treatment is partially rescued by apoptosis and autophagy inhibitors. (A–B) Annexin V/PI co-staining flow cytometry analysis. After treatment, the cells were collected and double-stained with FITC-conjugated Annexin V and PI. The analyses were performed with flow cytometry. Data are presented as the mean±SEM and were analyzed with a one-way ANOVA. *P<0.05, **P<0.01. (A) HCT116 cells were treated with OC alone (7.5 μmol/L), GUTK alone (7.5 μmol/L), or OC+GUTK (7.5 μmol/L each) for 48 h in complete medium, with or without pre-treatment with Z-VAD-fmk (40 μmol/L) or HCQ (5 μmol/L) for 0.5 h. (B) HCT-116 cells were treated with OC alone (2 μmol/L), GUTK alone (2 μmol/L), or OC+GUTK (2 μmol/L each) for 3 h in EBSS, with or without pre-treatment with Z-VAD-fmk (40 μmol/L) or HCQ (5 μmol/L) for 0.5 h.
OC and GUTK in combination promote ROS generation and JNK activation in HCT116 cells
To examine whether the combination of OC and GUTK promotes the generation of ROS in HCT116 colon cancer cells, we measured the ROS levels under a fluorescence microscope by using DCFH-DA staining. HCT116 cells were treated with OC, GUTK, or both compounds in combination in complete medium for 24 h (Figure 6A) or in EBSS medium for 3 h (Figure 6B). Combined treatment with OC and GUTK induced increases of ROS in both complete and EBSS medium. Pretreatment with the ROS scavenger N-acetylcysteine (NAC) abolished this increase in ROS. In addition, we explored the ability of this combination treatment to activate JNK. Specifically, OC and GUTK in combination increased the expression of phosphorylated JNK both in complete medium and in EBSS medium, without changing total JNK expression (Figure 6C, 6D). In EBSS medium, combined treatment with OC and GUTK increased JNK activation in a time-dependent manner (Figure 6E). To understand the connection between ROS, the JNK pathway and apoptosis, HCT116 cells were treated with a JNK inhibitor (SP600125) or a ROS scavenger (NAC) before treatment with OC and GUTK. Pretreatment with SP600125 or NAC for 0.5 h did not decrease the percentage of sub-G1 cells induced by the OC and GUTK combination treatment (Supplementary Figure S3A, S3B). These results suggested that the OC and GUTK combination treatment activated cell death not only via the ROS and JNK pathways but also via other mechanisms, which require further investigation.

OC and GUTK in combination promote ROS generation and JNK activation in HCT116 cells. (A, B) HCT116 cells were treated with OC or GUTK alone or both in combination, with or without pretreatment with N-acetylcysteine (NAC) (10 mmol/L) for 0.5 h. The cells were then treated with DCFH-DA (20 μmol/L) for 0.5 h and observed under a fluorescence microscope. (A) HCT116 cells were treated with OC alone (7.5 μmol/L), GUTK alone (7.5 μmol/L), or OC+GUTK (7.5 μmol/L each) for 48 h in complete medium. (B) HCT-116 cells were treated with OC alone (2 μmol/L), GUTK alone (2 μmol/L) or OC+GUTK (2 μmol/L) for 3 h in EBSS. Scale bar, 20 μm. (C–E) The expression of phospho-JNK was detected by Western blotting. Tubulin was used as a loading control. (C) HCT116 cells were treated with OC or GUTK alone (1, 2, 5, and 7.5 μmol/L) or OC combined with GUTK (1, 2, 5, and 7.5 μmol/L) for 48 h in complete medium. (D) HCT116 cells were treated with OC or GUTK alone (1, 2, and 5 μmol/L) or OC combined with GUTK (1, 2, and 5 μmol/L) for 3 h in EBSS. (E) HCT116 cells were treated with OC alone (2 μmol/L), GUTK alone (2 μmol/L) or OC+GUTK (2 μmol/L each) for 3, 6, or 9 h in EBSS.
Discussion
G yunnanensis contains up to 5% OC and GUTK. These compounds are structurally similar, but their anticancer mechanisms differ. Specifically, OC exhibits multiple anticancer activities, including promoting apoptosis and inhibiting autophagy and metastasis15,16,17, whereas GUTK has been shown to promote apoptosis, arrest the cell cycle, and promote autophagy18,19. At low doses, neither OC nor GUTK alone affected HCT116 cells, whereas combined treatment with OC and GUTK at the same doses produced potent effects, including increasing the sub-G1 population, cleavage and activation of casapse-3 and PARP cleavage. In addition, we found that the combination treatment also increased the LC3-IIB protein levels and decreased the p62 protein levels, thus suggesting that this treatment activated the autophagy pathway. These results indicated that OC and GUTK at their natural concentrations (1:1 molar ratio) exerted favorable addictive effects on HCT116 cells. Interestingly, the combination of OC and GUTK did not exert additive effects at other ratios (eg, 1:2 or 1:1.5) (Data not shown).
Plants contain many bioactive compounds that may functionally interact, thereby targeting multiple molecular processes and increasing therapeutic efficacy. For instance, EGb 761 is a standardized natural extract of Ginkgo biloba that contains 24% ginkgoflavonol glycosides and 6% terpene lactones, and ginkgoflavonol glycosides and terpene lactones are considered to be the main active components of Ginko biloba22. EGb 761 is currently prescribed for the treatment of cardiovascular disease in European countries23. Mixtures of green tea catechins with ointment have been shown to exhibit antitumor properties, and these formulations have been approved by the US Food and Drug Administration (FDA) to treat genital warts caused by human papilloma virus24. Moreover, the US FDA has published a botanical drug product guide based on multi-component plant extracts25. In addition, the fruit of G yunnanensis is is edible and has a pleasant sweet-and-sour taste26, which suggests that compounds derived from this fruit will be safe for human consumption. Nevertheless, future work, such as standardization, quality control, and pharmacokinetic and pharmacodynamics studies of G yunnanensis extracts, will be required to develop G yunnanensis as a potential anticancer drug.
Author contribution
Hong-xi XU and Yuan-zhi LAO designed the research; Hui LI, Li ZHANG, Bao-jun ZHANG, and Xin-yu LIU performed the experiments; Hong-sheng TAN and Wen-wei FU contributed new reagents or analytic tools; Hui LI, Yuan-zhi LAO, and Xiao-xiao MENG analyzed data and wrote the paper.
References
Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 2015; 136: E359–86.
DeSantis CE, Lin CC, Mariotto AB, Siegel RL, Stein KD, Kramer JL, et al. Cancer treatment and survivorship statistics, 2014. CA Cancer J Clin 2014; 64: 252–71.
Liu EY, Ryan KM . Autophagy and cancer — issues we need to digest. J Cell Sci 2012; 125: 2349–58.
Ouyang L, Shi Z, Zhao S, Wang FT, Zhou TT, Liu B, et al. Programmed cell death pathways in cancer: a review of apoptosis, autophagy and programmed necrosis. Cell Prolif 2012; 45: 487–98.
Farooqi AA, Fayyaz S, Hou MF, Li KT, Tang JY, Chang HW . Reactive oxygen species and autophagy modulation in non-marine drugs and marine drugs. Mar Drugs 2014; 12: 5408–24.
Filomeni G, De Zio D, Cecconi F . Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ 2015; 22: 377–88.
Kim GT, Lee SH, Kim JI, Kim YM . Quercetin regulates the sestrin 2-AMPK-p38 MAPK signaling pathway and induces apoptosis by increasing the generation of intracellular ROS in a p53-independent manner. Int J Mol Med 2014; 33: 863–9.
Guo X, Chen S, Zhang Z, Dobrovolsky VN, Dial SL, Guo L, et al. Reactive oxygen species and c-Jun N-terminal kinases contribute to TEMPO-induced apoptosis in L5178Y cells. Chem Biol Interact 2015; 235: 27–36.
Romano B, Borrelli F, Pagano E, Cascio MG, Pertwee RG, Izzo AA . Inhibition of colon carcinogenesis by a standardized Cannabis sativa extract with high content of cannabidiol. Phytomedicine 2014; 21: 631–9.
Goldar S, Khaniani MS, Derakhshan SM, Baradaran B . Molecular mechanisms of apoptosis and roles in cancer development and treatment. Asian Pac J Cancer Prev 2015; 16: 2129–44.
Ovadje P, Roma A, Steckle M, Nicoletti L, Arnason JT, Pandey S . Advances in the research and development of natural health products as main stream cancer therapeutics. Evid Based Complement Alternat Med 2015; 2015: 751348. doi: 10.1155/2015/751348.
Wu SB, Long C, Kennelly EJ . Structural diversity and bioactivities of natural benzophenones. Nat Prod Rep 2014; 31: 1158–74.
Anholeti MC, Paiva SR, Figueiredo MR, Kaplan MA . Chemosystematic aspects of polyisoprenylated benzophenones from the genus Clusia. An Acad Bras Cienc 2015; 87: 289–301.
Xu G, Feng C, Zhou Y, Han QB, Qiao CF, Huang SX, et al. Bioassay and ultraperformance liquid chromatography/mass spectrometry guided isolation of apoptosis-inducing benzophenones and xanthone from the pericarp of Garcinia yunnanensis Hu. J Agric Food Chem 2008; 56: 11144–50.
Feng C, Zhou LY, Yu T, Xu G, Tian HL, Xu JJ, et al. A new anticancer compound, oblongifolin C, inhibits tumor growth and promotes apoptosis in HeLa cells through Bax activation. Int J Cancer 2012; 131: 1445–54.
Lao Y, Wan G, Liu Z, Wang X, Ruan P, Xu W, et al. The natural compound oblongifolin C inhibits autophagic flux and enhances antitumor efficacy of nutrient deprivation. Autophagy 2014; 10: 736–49.
Wang X, Lao Y, Xu N, Xi Z, Wu M, Wang H, et al. Oblongifolin C inhibits metastasis by up-regulating keratin 18 and tubulins. Sci Rep 2015; 5: 10293.
Wu M, Lao Y, Xu N, Wang X, Tan H, Fu W, et al. Guttiferone K induces autophagy and sensitizes cancer cells to nutrient stress-induced cell death. Phytomedicine 2015; 22: 902–10.
Kan WL, Yin C, Xu HX, Xu G, To KK, Cho CH, et al. Antitumor effects of novel compound, guttiferone K, on colon cancer by p21Waf1/Cip1-mediated G0/G1 cell cycle arrest and apoptosis. Int J Cancer 2013; 132: 707–16.
Chou TC . Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev 2006; 58: 621–81.
Chou TC . Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res 2010; 70: 440–6.
van Beek TA, Montoro P . Chemical analysis and quality control of Ginkgo biloba leaves, extracts, and phytopharmaceuticals. J Chromatogr A 2009; 1216: 2002–32.
Nash KM, Shah ZA . Current perspectives on the beneficial role of Ginkgo biloba in neurological and cerebrovascular disorders. Integr Med Insights 2015; 10: 1–9.
Date AA, Destache CJ . Natural polyphenols: potential in the prevention of sexually transmitted viral infections. Drug Discov Today 2016; 21: 333–41.
U.S Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research. Guidance for Industry: Botanical Drug Products 2004.
Wu ZY, Chen J . Flora of Yunnan. Beijing: Science Press; 1991.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No 81173485 and 81273403) and the Natural Science Foundation of Shanghai (No 14ZR1441300).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Supplementary information is available on the Acta Pharmacologica Sinica's web site.
Supplementary information
Supplementary Figure S1
Among the fractions, GYFB (OC+GUTK fraction) potently inhibited HCT116 cells in a dose-dependent manner, where as GYFA (NO OC+GUTK fraction) exerted almost no effect on cell viability using a SYBR green assay. (JPG 402 kb)
Supplementary Figure S2
OC, GUTK and their combination (1:1 molar ratio) significantly decreased the viability of HCT116 cells in a dose-dependent manner according to LDH assays. (JPG 100 kb)
Supplementary Figure S3
Pretreatment with SP600125 or NAC for 0.5 h did not decrease the percentage of sub-G1 cells induced by the OC and GUTK combination treatment. (JPG 603 kb)
Rights and permissions
About this article

Cite this article
Li, H., Meng, Xx., Zhang, L. et al. Oblongifolin C and guttiferone K extracted from Garcinia yunnanensis fruit synergistically induce apoptosis in human colorectal cancer cells in vitro. Acta Pharmacol Sin 38, 252–263 (2017). https://doi.org/10.1038/aps.2016.101
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2016.101
Keywords
This article is cited by
-
Anticancer Properties and Mechanism of Action of Oblongifolin C, Guttiferone K and Related Polyprenylated Acylphloroglucinols
Natural Products and Bioprospecting (2021)

{kind=link}
{kind=link}
{kind=link}