
Abstract
Aim:
Berberine (BBR), an isoquinoline-derived alkaloid isolated from Rhizoma coptidis, exerts cardioprotective effects. Because endoplasmic reticulum (ER) stress plays a pivotal role in myocardial ischemia/reperfusion (MI/R)-induced apoptosis, it was interesting to examine whether the protective effects of BBR resulted from modulating ER stress levels during MI/R injury, and to define the signaling mechanisms in this process.
Methods:
Male rats were treated with BBR (200 mg·kg−1·d−1, ig) for 2 weeks, and then subjected to MI/R surgery. Cardiac dimensions and function were assessed using echocardiography. Myocardial infarct size and apoptosis was examined. Total serum LDH levels and CK activities, superoxide production, MDA levels and the antioxidant SOD activities in heart tissue were determined. An in vitro study was performed on cultured rat embryonic myocardium-derived cells H9C2 exposed to simulated ischemia/reperfusion (SIR). The expression of apoptotic, ER stress-related and signaling proteins were assessed using Western blot analyses.
Results:
Pretreatment with BBR significantly reduced MI/R-induced myocardial infarct size, improved cardiac function, and suppressed myocardial apoptosis and oxidative damage. Furthermore, pretreatment with BBR suppressed MI/R-induced ER stress, evidenced by down-regulating the phosphorylation levels of myocardial PERK and eIF2α and the expression of ATF4 and CHOP in heart tissues. Pretreatment with BBR also activated the JAK2/STAT3 signaling pathway in heart tissues, and co-treatment with AG490, a specific JAK2/STAT3 inhibitor, blocked not only the protective effects of BBR, but also the inhibition of BBR on MI/R-induced ER stress. In H9C2 cells, treatment with BBR (50 μmol/L) markedly reduced SIR-induced cell apoptosis, oxidative stress and ER stress, which were abolished by transfection with JAK2 siRNA.
Conclusion:
BBR ameliorates MI/R injury in rats by activating the AK2/STAT3 signaling pathway and attenuating ER stress-induced apoptosis.
Similar content being viewed by others


Introduction
Ischemic heart disease remains the leading cause of mortality and disability worldwide. Although timely reperfusion therapy is the primary treatment, reperfusion itself results in major cardiac damage, commonly referred to as myocardial ischemia/reperfusion (MI/R) injury1,2. The endoplasmic reticulum (ER) is a multifunctional organelle in eukaryotic cells, which participates in the biosynthesis and folding of proteins. MI/R injury is known to result in the accumulation of unfolded proteins in the ER, which causes a severe unfolded protein response (UPR). Persistent UPR eventually leads to cellular apoptosis3. Therefore, reducing excessive UPR, also referred to as ER stress, is of great importance in ameliorating MI/R injury.
Berberine (BBR, Figure 1), is an isoquinoline-derived alkaloid isolated from Rhizoma coptidis, which has been widely used clinically owing to its multiple biochemical and pharmacological effects4,5. Intriguingly, numerous clinical trials and animal studies have demonstrated the beneficial effects of BBR on cardiovascular performance6,7,8,9,10. BBR has been found to prevent ischemia-induced ventricular tachyarrhythmias, enhance the force of cardiac contractions, and decrease peripheral vascular resistance and blood pressure11,12,13,14. Recently, we have also demonstrated that Notch1 signaling plays a key role in BBR's cardioprotective action15. Interestingly, another recent study by Li et al. reported that BBR reduced myocardial ER stress in a rat model of cardiac hypertrophy through an autophagy-dependent mechanism12. However, to the best of our knowledge, it is still unknown whether BBR attenuates the ER stress caused by MI/R injury.

Chemical structure of berberine (BBR). Molecular weight: 336. Molecular formula: C20H18NO4+.
The Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway transduces cellular signals from the plasma membrane to the nucleus and plays a vital role in mediating cardioprotection against ischemia/reperfusion injury16,17,18. Importantly, JAK2/STAT3 signaling has been specifically proven to protect against MI/R injury17,19,20. In our previous experiments, activation of JAK2/STAT3 signaling by curcumin exerted a profound anti-oxidative effect against MI/R injury21. However, whether JAK2/STAT3 signaling can modulate myocardial ER stress level during MI/R injury has rarely been investigated. Furthermore, other studies have reported that BBR exerts a potential anti-tumor effect by modulating JAK2/STAT3 signaling22. BBR has also been shown to suppress IgE production by human B cells and peripheral blood mononuclear cells in food-allergic patients via STAT3 and T-box transcription factor TBX21 signaling23. It would therefore be interesting to explore whether BBR's cardioprotective action is associated with JAK2/STAT3 signaling during MI/R injury and its relationship with myocardial ER stress.
In the article, BBR was used as an adjuvant to attenuate the ER stress and oxidative stress induced by MI/R injury. In addition, the involvement of the JAK2/STAT3 signaling pathway in BBR's protective actions was also evaluated.
Materials and methods
Animals
Male Sprague-Dawley rats (200–250 g) were purchased from the Experimental Animal Center of the Fourth Military Medical University, China. The animals were housed under standard laboratory conditions (25±1 °C, 60% humidity, with a 12 h photoperiod) and provided free access to sterile food and water. All of the procedures were approved by the Committee of Experimental Animals of the Fourth Military Medical University and conformed to the Guide for the Care and Use of Laboratory Animals published by the United States National Institutes of Health (NIH Publication No 85-23, revised 1996).
Materials
BBR, sodium carboxymethylcellulose (CMC-Na), Evans blue and triphenyltetrazolium chloride (TTC) were purchased from Solarbio Technology (Beijing, China). A terminal deoxynucleotidyltransferase-mediated dUTP nick-end labeling (TUNEL) assay kit and a protease inhibitor cocktail were obtained from Roche Molecular Biochemicals (Mannheim, Germany). 4', 6-diamino-2-phenylindole (DAPI) was purchased from Sigma-Aldrich (St Louis, MO, USA). Lactate dehydrogenase (LDH), creatine kinase (CK), malondialdehyde (MDA), superoxide dismutase (SOD) and superoxide generation assay kits were purchased from the Institute of Jiancheng Bioengineering (Nanjing, China). A bicinchoninic acid (BCA) protein quantification kit was purchased from Merck Millipore Technology (Darmstadt, Germany). The primary antibodies against JAK2, p-JAK2 (Tyr1007/1008), STAT3, p-STAT3 (Try705), p-PERK (Thr981), PERK, p-eIF2α (Ser52), eIF2α, ATF4, CHOP, gp91phox, caspase-3, Bcl-2, Bax, and β-actin, AG490 (the specific inhibitor of JAK2/STAT3 signaling), and JAK2 siRNA were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Rabbit anti-goat, goat anti-rabbit and goat anti-mouse secondary antibodies were purchased from the Zhongshan Company (Beijing, China).
Study groups and experimental protocol
First, we evaluated the toxic effects of BBR and AG490 on the sham-operated rat hearts. We hypothesized that under the present experimental dosage, BBR or AG490 treatment would have no significant toxic effects on the sham-operated rat hearts. The experiments were conducted as follows (n=8): 1) Sham group: normal rats were only subjected to the sham operation; 2) Sham+BBR group: the rats were orally treated with BBR (in 0.5% CMC-Na solution, 200 mg·kg−1·d−1, 2 weeks) and then subjected to the sham operation; 3) Sham+AG group: the rats were treated with AG490 (5 mg·kg−1·d−1, ip, 3 days before MI/R operation, 20 mg/kg, 10 min after the beginning of the operation) and then subjected to the sham operation. The doses of BBR and AG490 were selected based on previous reports24,25,26,27.
Second, we tested the effects of BBR and AG490 on the MI/R-injured rat hearts. The experiments were conducted as follows (n=8): 1) MI/R+V group: normal rats were orally gavaged with 0.5% CMC-Na solution (2 mL/d) for 2 weeks and then subjected to the MI/R operation; 2)MI/R+BBR group: normal rats were orally treated with BBR as described above and then subjected to the MI/R operation; 3) MI/R+BBR+AG: normal rats were treated with BBR as well as AG490 as described above, and then subjected to the MI/R operation; 4) MI/R+AG: normal rats were treated with AG490 alone as described before and then subjected to the MI/R operation.
Third, we determined the role of JAK2/STAT3 signaling in BBR's protective effect in cultured H9C2 cells. We chose 50 μmol/L as the experimental concentration based on our preliminary experiment and the previous reports15,24. After preparation, the cells were divided into the following groups (8 separate experiments were performed for each experimental condition): 1) Simulated ischemia/reperfusion (SIR) group: the cardiomyocytes were incubated in normal DMEM for 28 h, subjected to simulated ischemia for 2 h, and then incubated in normal DMEM for 4 h to simulate reperfusion; 2) SIR+BBR group: the cardiomyocytes were incubated in normal DMEM for 24 h, incubated in DMEM with 50 μmol/L BBR for 4 h, and then subjected to the SIR treatment; 3) SIR+BBR+JAK2 siRNA group: after the cells were transfected with JAK2 siRNA, they were treated with 50 μmol/L BBR for 4 h and then exposed to SIR injury. 4) SIR+JAK2 siRNA: the cardiomyocytes were transfected with JAK2 siRNA and then exposed to the SIR injury.
MI/R surgery
The MI/R surgery was performed as described previously with modifications28. Briefly, after the rats were anesthetized with a 3% pentobarbital sodium, myocardial ischemia was induced by exteriorizing the heart through a left thoracic incision, placing a 6–0 silk suture around the left anterior descending (LAD) coronary artery and tying a slipknot. After 30 min of ischemia, the slipknot was released, and the myocardium was reperfused for 4 h (for analysis of protein expression), 6 h (for quantification of myocardial apoptosis and infarct size) or 24 h (for the determination of cardiac function recovery). The sham group was subjected to the same operation procedures except that the suture passed under the left coronary artery was left untied.
Cardiac function measurement
Echocardiography was performed 24 h after reperfusion as previously described29. Briefly, the rats were sedated (2% isoflurane) and studied using a VEVO 770 high-resolution in vivo imaging system (Visual Sonics, Toronto, Canada). The cardiac dimensions and function were assessed using M-mode echocardiography. The LV end-diastolic diameter and LV end-systolic diameter were measured on the parasternal LV long-axis view. All measurements represent the mean of 5 consecutive cardiac cycles. The left ventricular ejection fraction (LVEF) and left ventricular fractional shortening (LVFS) were calculated using computer algorithms. All of these measurements were performed in a blinded manner.
Myocardial infarct size measurement
The slipknot around the LAD coronary artery was retied at the end of the reperfusion, and 1 mL of 1% Evans blue dye was injected into the aortic artery. The heart was then quickly excised and frozen at −80 °C. After that, the frozen heart was sliced transversally into 1-mm thick sections and then incubated at 37 °C for 30 min in 2% TTC solution as described in our previous studies29. Then, digital images were captured and analyzed. The sizes of the areas of infarct (INF) and areas at risk (AAR) were measured digitally by using Image-Pro Plus software (Media Cybernetics, Bethesda, MD, USA). The INF and AAR were expressed as percentages of the LV area (INF/LV and AAR/LV, respectively).
Myocardial apoptosis measurement
After the reperfusion, the hearts were fixed in 4% paraformaldehyde in PBS (pH 7.4) for 24 h at room temperature. The fixed tissues were then embedded in paraffin, and TUNEL staining was performed according to the manufacturer's instructions as described in our previous studies29. All nuclei were stained by DAPI. The apoptotic index (ie, the number of TUNEL-positive nuclei/total number of nuclei counted × 100%) was determined in a blinded manner.
Determination of total serum LDH and CK
After the end of the reperfusion, blood samples (0.5 mL) were drawn and total serum lactate dehydrogenase (LDH) and creatine kinase (CK) activities were measured spectrophotometrically (Beckman DU 640, Fullerton, CA, USA) in a blinded manner as described before29. All kits for measuring LDH and CK activities were purchased from the Institute of Jiancheng Bioengineering (Nanjing, China).
Oxidative damage measurement
Superoxide production in the tissues and cells was measured using lucigenin-enhanced chemiluminescence, as described previously30. Superoxide production was expressed as relative light units (RLU) per second per milligram heart weight (RLU·mg−1·s−1). The MDA levels and the activities of the antioxidant SOD in the heart homogenates were determined spectrophotometrically, as previously described30.
Cell culture and siRNA transfection
H9C2 embryonic rat myocardium-derived cells (Shanghai Tiancheng Technology Co, Ltd), a well-characterized cell line used to study myocardial cell ischemia, were grown in Dulbecco's modified Eagle's medium (DMEM, GIBCO) supplemented with 10% inactivated fetal bovine serum (FBS, GIBCO), 50 U/mL penicillin and 50 μg/mL streptomycin (GIBCO) at 37 °C in a humidified atmosphere with 5% CO2. The siRNA transfection was carried out according to the manufacturer's instructions. Briefly, 2×105 H9C2 cells were seeded in a six-well culture plate in 2 mL of antibiotic-free normal growth medium supplemented with FBS. After preparation of the mixture of the siRNA duplex solution and transfection reagent, the siRNA transfection medium and the siRNA transfection reagent mixture were added to the cells. After 6 h of incubation, 1 mL of normal growth medium containing twice the normal serum concentration was added without removing the transfection mixture. Then, the cells were incubated for an additional 18 h.
SIR treatment
The SIR treatment was performed using physiological concentrations of potassium, hydrogen, and lactate. The procedure was performed as described previously15. Briefly, the H9C2 cells were exposed to an ischemic buffer containing (in mmol/L) 137 NaCl, 12 KCl, 0.49 MgCl2, 0.9 CaCl2, and 4 HEPES. This buffer was also supplemented with (in mmol/L) 10 deoxyglucose, 0.75 sodium dithionate, and 20 lactate. The buffer pH was 6.5. The cardiomyocytes were incubated for 2 h in a humidified cell culture incubator (21% O2, 5% CO2, 37 °C). Reperfusion was performed by returning the cells to normal culture medium for 4 h in a humidified cell culture incubator (21% O2, 5% CO2, 37 °C).
Cell viability measurement
The H9C2 cells, 1×104 per well, were seeded in 96-well culture plates. The cell viability was measured using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay as described previously15. Briefly, after the cells were treated and washed with PBS, 10 μL of MTT dye was added to each well at a final concentration of 0.5 mg/mL. After 4 h of reperfusion, 100 μL of DMSO was added to dissolve the formazan crystals. The absorbance was measured using a microtiter plate reader (SpectraMax 190, Molecular Device, USA) at a wavelength of 570 nm. The cell viability was calculated by dividing the optical density of samples by the optical density of the control group.
Western blot evaluation
The proteins from the rat hearts and cultured cells were extracted in RIPA buffer [20 mmol/L Tris-HCl (pH 7.4), 150 mmol/L sodium chloride, 2 mmol/L EGTA, 0.5% sodium deoxycholate, 1% Triton-X 100, 100 mmol/L sodium fluoride, and 2 mmol/L sodium orthovanadate] containing protease inhibitors. The concentration of the protein extracted from each specimen was quantified using the BCA protein assay kit. After separating the proteins by SDS-PAGE, they were transferred to PVDF membranes (Millipore, MA, USA). The membranes were probed with the primary antibodies against JAK2, p-JAK2, STAT3, p-STAT3, PERK, p-PERK, p-eIF2α, eIF2α, ATF4, CHOP, gp91phox, caspase-3, Bcl-2, Bax or β-actin (1:500 in TBST) overnight at 4 °C. After washing three times with TBST, the membranes were incubated with the secondary antibody in TBST for 2 h at 37 °C, then washed as described above. The positive protein bands were developed using a chemiluminescence system, and the bands were scanned and quantified by densitometric analysis using an image analyzer Quantity One System (Bio-Rad, Richmond, CA, USA).
Statistical analysis
The data are presented as the mean±SEM. The statistical analyses were performed using a 2-tailed Student t-test for unpaired observations or a one-way ANOVA followed by the Bonferroni post hoc test for multiple comparisons. A P<0.05 was considered statistically significant.
Results
Exogenous BBR treatment significantly attenuated the MI/R injury and oxidative damage, whereas these effects were abolished by AG490 co-treatment
In the in vivo experiment, we first evaluated the effect of exogenous BBR or AG490 treatment on the sham-operated rat hearts. Compared with the sham group, neither BBR nor AG490 treatment significantly affected the cardiac function. As seen in Figure 2A–2C, neither BBR nor AG490 had any significant effect on the LVEF and LVFS (compared with the sham group, P>0.05). Similarly, neither the myocardial infarct size nor the myocardial apoptosis were markedly affected by treatment with either BBR or AG490 (compared with the Sham group, P>0.05, Figure 2D–2G). Moreover, we evaluated the effects of AG or BBR on the cardiac JAK2/STAT3 signaling. As shown in Figure 2H–2J, although AG treatment markedly inhibited the myocardial JAK2/STAT3 signaling (P<0.01), BBR alone had no significant effect (P>0.05).

BBR or AG490 treatment had no significant toxic effect on sham-operated rat hearts. Normal rats pretreated with BBR or AG490 were subjected to the sham operation. After 24 h of reperfusion, echocardiography was performed. After 6 h of reperfusion, the myocardial infarct size and apoptosis index were evaluated. (A) Representative M-mode echocardiographic images. (B) Left ventricular ejection fraction (LVEF). (C) Left ventricular fractional shortening (LVFS). (D) Representative heart section images. The Evans blue-stained areas (blue) indicate the non-ischemic/reperfused area; the TTC-stained areas (red) indicate ischemic but viable tissue; and the Evans blue/TTC-unstained (negative) areas (white) indicate infarcted myocardium. (E) Representative images of apoptotic cardiomyocytes by TUNEL staining. The green fluorescence shows the TUNEL-positive nuclei; the blue fluorescence shows the nuclei of all cardiomyocytes; original magnification, ×400. (F) The myocardial infarct size expressed as the percentage of area-at-risk. (G) Percentage of TUNEL-positive nuclei. (H) Representative blots. (I) p-JAK2/JAK2 ratio. (J) p-STAT3/STAT3 ratio. BBR, berberine; AG, AG490. The results are expressed as the mean±SEM. n=8/group. cP<0.01 vs the sham group. fP<0.01 vs the sham+BBR group.
Next, we found that the BBR treatment effectively improved the post-MI/R cardiac function recovery as indicated by the increased LVEF and LVFS (compared with the MI/R+V group, P<0.01, Figure 3A–3C). However, compared with the MI/R+BBR group, these protective effects were blocked by AG490 co-treatment (P<0.01, Figure 3A–3C). AG490 alone did not significantly affect the cardiac function compared with the MI/R+V group (P>0.05, Figure 3A–3C). We also measured the levels of myocardial apoptosis. As shown in Figure 3D and 3E, the myocardial apoptosis caused by the MI/R injury was also markedly attenuated by BBR treatment, and this attenuation was also abolished by the AG490 co-treatment. Similarly, we did not observe a significant difference between the MI/R+AG and MI/R+V groups (P>0.05, Figure 3D and 3E). Next, we evaluated the myocardial infarct sizes. As shown in Figure 3F and 3G, the BBR-treated group showed a significantly decreased myocardial infarct size (compared with the MI/R+V group, P<0.01), while the AG490 treatment significantly blocked this effect (compared with the MI/R+BBR group, P<0.01). We also did not observe a significant difference between the MI/R+AG and MI/R+V groups (P>0.05, Figure 3F and 3G). The BBR treatment significantly decreased the serum LDH and CK levels, and these effects were also blocked by AG490 treatment (P<0.01, Figure 4A and 4B).

BBR effectively reduced the MI/R injury, whereas AG490 treatment blocked this effect. Normal rats pretreated with BBR or AG490 were subjected to the MI/R operation. After 24 h of reperfusion, echocardiography was performed. After 6 h of reperfusion, the myocardial infarct size and apoptotic index were evaluated. (A) Representative M-mode echocardiographic images. (B) Left ventricular ejection fraction (LVEF). (C) Left ventricular fractional shortening (LVFS). (D) Representative images of apoptotic cardiomyocytes by TUNEL staining. The green fluorescence shows the TUNEL-positive nuclei; the blue fluorescence shows the nuclei of all cardiomyocytes; original magnification, ×400. (E) Percentage of TUNEL-positive nuclei. (F) Representative heart section images. The Evans blue-stained areas (blue) indicate the non-ischemic/reperfused area; the TTC-stained areas (red) indicate ischemic but viable tissue; and the Evans blue/TTC-unstained (negative) areas (white) indicate infarcted myocardium. (G) The myocardial infarct size expressed as the percentage of area-at-risk. BBR, berberine; AG, AG490. The results are expressed as the mean±SEM. n=8/group. cP<0.01 vs the MI/R+V group; fP<0.01 vs the MI/R+BBR group.

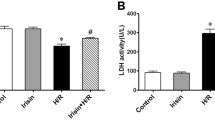
BBR effectively ameliorated cardiac necrosis and oxidative stress, whereas AG490 treatment blocked these effects. Normal rats treated with BBR or AG490 were subjected to the MI/R operation. All measurements were performed after 6 h of reperfusion. (A) Serum LDH levels. (B) Serum CK levels. (C) Cardiac superoxide generation. (D) gp91phox expression. Top images: representative blots. (E) Myocardial MDA contents. (F) Myocardial SOD contents. BBR, berberine; AG, AG490. The results are expressed as the mean±SEM. n=8/group. cP<0.01 vs the MI/R+V group; fP<0.01 vs the MI/R+BBR group; iP<0.01 vs the MI/R+BBR+AG group.
Furthermore, we evaluated the oxidative damage caused by the MI/R injury. As shown in Figure 4C,4E and 4F, the BBR treatment significantly decreased the myocardial superoxide production and MDA levels and increased the myocardial SOD activity (P<0.01, compared with the MI/R+V group). In addition, we measured the expression of gp91phox, a major component of NADPH oxidase. As shown in Figure 4D, the MI/R injury significantly up-regulated the gp91phox expression, but this was markedly down-regulated by the BBR treatment. However, all the protective effects of BBR were abolished by the AG490 co-treatment (compared with the MI/R+BBR group, P<0.01, Figure 4C–4F). Consistent with the other results, AG490 alone did not cause a significant difference in the myocardial oxidative damage compared with the MI/R+V group (P>0.05, Figure 4C–4F).
BBR treatment reduced cardiac apoptosis in MI/R hearts via JAK2/STAT3 signaling pathway
To further understand the molecular mechanism of BBR's cardioprotective action, we investigated the myocardial JAK2/STAT3 signaling pathway. Figure 5B–5C showed that BBR markedly up-regulated the p-JAK2 and p-STAT3 levels in the MI/R-injured hearts (compared with the MI/R+V group, P<0.01). However, these effects were blocked by the AG490 treatment (compared with the MI/R+BBR group, P<0.01, Figure 5B and 5C). In addition, BBR also significantly attenuated the apoptosis caused by the MI/R injury as indicated by markedly decreased levels of caspase-3 and Bax and an increased level of Bcl-2 (compared with the MI/R+V group, P<0.01, Figure 5D–5F). As expected, AG490 also blocked these effects by increasing the expression of caspase-3 and Bax and decreasing the expression of Bcl-2 (compared with the MI/R+BBR group, P<0.01, Figure 5D–5F). Although AG490 alone down-regulated the p-JAK2 and p-STAT3 levels, it did not significantly aggravate the myocardial apoptosis (compared with the MI/R+V group, P>0.05, Figure 5D–5F).

BBR up-regulated cardiac JAK2/STAT3 signaling and down-regulated myocardial apoptosis, whereas AG490 treatment blocked these effects. Normal rats pretreated with BBR or AG490 were subjected to the MI/R operation. All measurements were performed after 4 h of reperfusion. (A) Representative blots. (B) p-JAK2/JAK2 ratio. (C) p-STAT3/STAT3 ratio. (D) Caspase-3 expression. (E) Bcl-2 expression. (F) Bax expression. BBR, berberine; AG, AG490. The results are expressed as the mean±SEM. n= 8/group. cP<0.01 vs the MI/R+V group; fP<0.01 vs the MI/R+BBR group; iP<0.01 vs the MI/R+BBR+AG group.
Exogenous BBR treatment significantly attenuated the MI/R injury-induced ER stress, but this effect was abolished by AG490 co-treatment
We next measured the PERK/eIF2α/ATF4-mediated ER stress levels in the hearts. As shown in Figure 6, the BBR treatment significantly attenuated the MI/R-induced myocardial ER stress level by down-regulating the myocardial PERK and eIF2α phosphorylation levels and the expression of ATF4 and CHOP (compared with the MI/R+V group, P<0.01). In addition, these effects were blocked by AG490 treatment (compared with the MI/R+BBR group, P<0.01). AG490 alone did not have a significant effect on the myocardial ER stress level compared with the MI/R+V group (P>0.05, Figure 6).

BBR significantly attenuated the PERK/eIF2α/ATF4-mediated myocardial ER stress, whereas AG490 treatment blocked this effect. Normal rats pretreated with BBR or AG490 were subjected to the MI/R operation. Western blotting analysis was performed after 4 h of reperfusion. (A) Representative blots; (B) p-PERK/PERK ratio; (C) p-eIF2α/eIF2α ratio; (D) ATF4 expression; (E) CHOP expression. BBR, berberine; AG, AG490. The results are expressed as the mean±SEM. n=8/group. cP<0.01 vs the MI/R+V group; eP<0.05, fP<0.01 vs the MI/R+BBR group; iP<0.01 vs the MI/R+BBR+AG group.
BBR inhibited the oxidative stress and protected the H9C2 cells from the SIR-induced apoptosis and oxidative damage, but these effects were abolished by JAK2 siRNA
The in vitro experimental results were consistent with those obtained from the MI/R-injured rat hearts. The H9C2 cells were first treated with BBR at 0.5, 5, 50, or 500 μmol/L for 4 h, and then the cell viability assay was performed. We did not observe significant cell viability changes in the 0.5, 5 and 50 μmol/L groups compared with the control group (P>0.05, Figure 7A). However, the 500 μmol/L BBR treatment markedly affected cell survival (P<0.01, compared with the Con group, Figure 7A). Therefore, the 50 μmol/L BBR dose was chosen for the present study. Second, we found that the BBR treatment effectively increased the cell viability following SIR (compared with SIR group, P<0.01, Figure 7B). Furthermore, the BBR treatment markedly decreased the apoptotic index (compared with the SIR group, P<0.01, Figure 7C and 7D). However, the protective action of BBR was abolished by treatment with the JAK2 siRNA (compared with SIR+BBR group, P<0.01). Significantly reduced levels of superoxide generation and gp91phox were found in the SIR+BBR group (compared with the SIR group, P<0.01, Figure 8A and 8B). However, the JAK2 siRNA treatment nearly abolished these effects (P<0.01, Figure 8A and 8B). The JAK2 siRNA had little effect on the cell viability, apoptotic index and oxidative damage compared with the SIR group (P>0.05, Figures 7 and 8).

In the cultured H9C2 cells subjected to SIR treatment, BBR significantly reduced the cellular apoptosis, whereas JAK2 siRNA markedly blocked these effects. H9C2 cells were exposed to BBR treatment for 4 h at the concentrations of 0.5, 5, 50, or 500 μmol/L. Then, cell viability was measured using MTT. Then, the cells were exposed to the SIR treatment with or without BBR or JAK2 siRNA treatment. (A) The cardiomyocyte viability was calculated by dividing the optical density of samples by the optical density of sham control. (B) The cardiomyocyte viability was evaluated using MTT. (C) Representative photomicrographs of TUNEL staining. The green fluorescence shows the TUNEL-positive nuclei; the blue fluorescence shows the nuclei of all cardiomyocytes; original magnification, ×200. (D) Percentage of TUNEL-positive nuclei. The results are expressed as the mean±SEM of 8 separate experiments with triplicate samples for each experimental condition within each experiment. cP<0.01 vs the control group; fP<0.01 vs the SIR group; iP<0.01 vs the SIR+BBR group.

In the cultured H9C2 cells subjected to the SIR treatment, BBR effectively reduced the oxidative stress, whereas the JAK2 siRNA blocked this effect. The cells were exposed to the SIR treatment with or without BBR or JAK2 siRNA treatment. Then, the cardiomyocyte oxidative stress markers were measured. (A) Cardiomyocyte superoxide generation. (B) gp91phox expression; top images: representative blots. The results are expressed as the mean±SEM of 8 separate experiments with triplicate samples for each experimental condition within each experiment. cP<0.01 vs the SIR group; fP<0.01 vs the SIR+BBR group; iP<0.01 vs the SIR+BBR+JAK2 siRNA group.
JAK2/STAT3 signaling contributed to BBR's protective effect on the H9C2 cells subjected to the SIR injury
BBR treatment significantly increased the expression of p-JAK2, p-STAT3, Bcl-2 and decreased that of caspase-3 and Bax (compared with the SIR group, P<0.01, Figure 9). However, these effects were also blocked by the JAK2 siRNA co-treatment (compared with the SIR+BBR group, P<0.01, Figure 9). Additionally, although the JAK2 siRNA treatment significantly decreased the expression of p-JAK2, p-STAT3, total-JAK2 and total-STAT3 (compared with the SIR group, P<0.01, Figure 9A–9C), it had little effect on the apoptosis-associated protein expression (compared with the SIR group, P<0.01, Figure 9D–9F).

In the cultured H9C2 cells subjected to the SIR treatment, BBR effectively up-regulated the cardiac JAK2/STAT3 signaling and down-regulated the myocardial apoptosis, whereas JAK2 siRNA blocked these effects. The cells were exposed to the SIR treatment with or without BBR or JAK2 siRNA treatment. Then, cardiomyocyte protein expression was measured. (A) Representative blots. (B) p-JAK2/JAK2 ratio. (C) p-STAT3/STAT3 ratio. (D) Caspase-3 expression. (E) Bcl-2 expression. (F) Bax expression. The results are expressed as the mean±SEM. n=8/group. cP<0.01 vs the SIR group; fP< 0.01 vs the SIR+BBR group; iP<0.01 vs the SIR+BBR+JAK2 siRNA group.
JAK2/STAT3 activation by the BBR treatment reduced the ER stress in the H9C2 cells subjected to SIR injury
Finally, we determined the effect of BBR on the PERK/eIF2α/ATF4-mediated ER stress level in the SIR-treated cardiomyocytes. Consistently, BBR treatment effectively reduced the cellular ER stress by decreasing the myocardial PERK and eIF2α phosphorylation levels and reducing the expression of ATF4 and CHOP (compared with the MI/R+V group, P<0.01, Figure 10). In addition, these effects were abolished by the AG490 co-treatment (compared with the MI/R+BBR group, P<0.01). The JAK2 siRNA alone did not significantly affect the cellular ER stress level compared with the MI/R+V group (P>0.05, Figure 10).

In the cultured H9C2 cells subjected to the SIR treatment, BBR significantly attenuated the PERK/eIF2α/ATF4-mediated myocardial ER stress, whereas JAK2 siRNA abolished this effect. (A) Representative blots. (B) p-PERK/PERK ratio. (C) p-eIF2α/eIF2α ratio. (D) ATF4 expression. (E) CHOP expression. The results are expressed as the mean±SEM. n=8/group. cP<0.01 vs the SIR group; fP<0.01 vs the SIR+BBR group; iP<0.01 vs the SIR+BBR+JAK2 siRNA group.
Discussion
In the present study, we utilized both in vitro and in vivo models to investigate the protective effect of BBR against MI/R-induced cardiac damage. We found that BBR markedly ameliorated the MI/R injury by reducing the PERK/eIF2α/ATF4-mediated ER stress. Importantly, JAK2/STAT3 signaling was proven to play a critical role in this process. This study could help to understand the pharmacology of BBR in the treatment of ischemic heart disease and promote the development of a novel strategy against reperfusion injury.
Recently, BBR has attracted extensive attention as a therapeutic agent against a number of diseases, including metabolic syndrome, fatty liver disease and coronary artery disease31,32,33. Importantly, the cardioprotective activity of BBR has been widely recognized24,34,35. In our recent study, both in vivo and in vitro experiments showed that BBR ameliorated MI/R injury by modulating Notch1/Hairy and enhancer of split 1 (Hes1) signaling15. The present study was carried out to further elucidate the underlying mechanisms of BBR's cardioprotective action. It has been proven that excessive ER stress participates in a number of pathological conditions, including ischemia/reperfusion injury, septic organ damage and diabetic complications36,37,38. Of the many signaling pathways involved in ER stress, PERK/eIF2α/ATF4 has been found to contribute strongly to programmed cell death during MI/R injury. Previously, Guo et aldemonstrated that PERK/eIF2α-mediated ER stress reduced the ischemic intolerance of diabetic hearts39. Moreover, hypoxic preconditioning was demonstrated to protect against hypoxia/reoxygenation (H/R) injury by attenuating the PERK/eIF2α-mediated ER stress40. Intriguingly, BBR has been proven to reduce ER stress in several pathological conditions. It has been found that BBR reduced the pro-inflammatory cytokine-induced ER stress in human intestinal epithelial cells41. Yu et al also demonstrated that BBR protected human renal proximal tubular cells from H/R injury by inhibiting the endoplasmic reticulum and mitochondrial stress pathways42. In the present study, we found that BBR decreased the phosphorylation levels of PERK and eIF2α and the expression of ATF4 and CHOP, thus reducing the ER stress caused by the MI/R injury. To the best of our knowledge, this is to determine the role of ER stress in BBR's cardioprotective action against MI/R injury. On the other hand, Endo et al. found that the ER stress pathway involving CHOP was activated and played a role in the pathogenesis of septic organ injury37. Interestingly, a recent study also demonstrated that CHOP inhibition effectively decreased the organ damage and increased the overall survival rate in septic mice43. Thus, it is worthwhile to further investigate BBR's potent therapeutic effect on septic organ injury with a focus on CHOP.
JAK/STAT signaling, the crucial intracellular signaling pathway linked to the activation of certain cytokine receptors, has been proven to mediate various cellular activities44,45. Upon activation, the JAKs phosphorylate certain cytoplasmic transcription factors, the STATs, which induces dimerization and translocation of the STATs to the nucleus where they regulate the expressions of specific genes. So far, four mammalian JAKs (JAK1, 2, 3, and Tyk2) and seven mammalian STATs (STAT1, 2, 3, 4, 5a, 5b, and 6) have been discovered46. Importantly, previous studies have specifically implicated JAK2/STAT3 signaling as having a key role in ameliorating the MI/R-induced cardiac injury17,47. It has been demonstrated that JAK2/STAT3 signaling mediates the cardioprotective effects of both ischemic preconditioning and postconditioning48,49. AG490, the inhibitor of JAK2/STAT3 signaling, resulted in the inhibition of STAT3 phosphorylation, enhanced cardiac apoptosis and abrogated the cardioprotective effects of preconditioning or postconditioning48,49. Of great interest, BBR modulated the ε-germline transcript expression in the peripheral blood mononuclear cells from food-allergic patients through STAT3 signaling23. In addition, BBR was also demonstrated to suppress the growth of several tumor cell lines including nasopharyngeal carcinoma cells, osteosarcoma cells, myeloma cells and prostate cancer cells through modulation of STAT3 signaling22,50,51,52. In this study, we found that BBR's cardioprotective effect was significantly blocked by AG490 or JAK2 siRNA, indicating that JAK2/STAT3 signaling also played a pivotal role under these conditions. Interestingly, a recent study by Li et alfound that Rho-kinase inhibition by fasudil improved the sarco/endoplasmic reticulum calcium ATPase (SERCA) activity and attenuated the activation of ER stress, possibly through JAK2/STAT3 signaling53. However, few studies have investigated the role of JAK2/STAT3 signaling in modulating the myocardial ER stress during MI/R injury. In the present study, we provided direct in vivo and in vitro evidence that JAK2/STAT3 activation by BBR reduced the cardiac ER stress level and that blocking this signaling significantly attenuated the ameliorative effect. Although further investigation is warranted to investigate the detailed molecular mechanisms, we did demonstrate that BBR reduced the myocardial ER stress by activating JAK2/STAT3 signaling.
Previously, Negoro et al. showed that activation of STAT3 protected cardiomyocytes from hypoxia/reoxygenation-induced oxidative stress by reducing the ROS-induced damage54. We have also demonstrated that JAK2/STAT3 activation attenuated the mitochondrial oxidative damage induced by MI/R injury55. Similarly, Mazen et al. reported that myocardial STAT3 signaling might also be modulated by cardiac redox status and attenuated by oxidative stress during heart failure and other acute or chronic cardiac diseases 56,57. Thus, intracellular STAT3 activity and oxidative stress might be bi-directionally regulated during the ischemia-reperfusion period. In this study, we showed that BBR treatment conferred a strong anti-oxidative effect against MI/R injury, whereas inhibition of JAK2/STAT3 signaling impeded this effect. Although the relationship between JAK2/STAT3 signaling and the cellular oxidative stress levels under these conditions needs to be further investigated, we proved that JAK2/STAT3 plays a key role in BBR's cardioprotective actions.
So far, several clinical investigations have established that there is a beneficial effect of BBR treatment on the cardiovascular system in patients with moderate hyperlipidemia and excess body weight7,58,59. Hence, BBR was suggested to be a safe and effective option for patients under conditions of moderate cardiovascular risk7. Based on our data, even short-term consumption of BBR might be a novel strategy to ameliorate the MI/R injury if it were administered several days before cardiac injury. Further clinical studies are needed before a conclusion can be reached on the utility of BBR in this setting.
Taken together, we demonstrated that BBR reduced myocardial ER stress via activation of JAK2/STAT3 signaling, thus ameliorating the MI/R injury. These findings may provide new mechanistic insight into the cardioprotective effects of berberine.
Author contribution
Yun WANG and Shi-qiang YU defined the research theme and revised the manuscript critically. Guo-long ZHAO, Li-ming YU and Wen-li GAO designed methods and experiments, carried out the laboratory experiments, and wrote the paper; Wei-xun DUAN, Bo JIANG, Xu-dong LIU, Bin ZHANG, Zhen-hua LIU, Meng-en ZHAI and Zhen-xiao JIN collected and analyzed the data and interpreted the results.
References
Elahi MM, Kong YX, Matata BM . Oxidative stress as a mediator of cardiovascular disease. Oxid Med Cell Longev 2009; 2: 259–69.
Kaminski KA, Bonda TA, Korecki J, Musial WJ . Oxidative stress and neutrophil activation–the two keystones of ischemia/reperfusion injury. Int J Cardiol 2002; 86: 41–59.
Xu J, Zhou Q, Xu W, Cai L . Endoplasmic reticulum stress and diabetic cardiomyopathy. Exp Diabetes Res 2012; 2012: 827971.
Stermitz FR, Lorenz P, Tawara JN, Zenewicz LA, Lewis K . Synergy in a medicinal plant: antimicrobial action of berberine potentiated by 5'-methoxyhydnocarpin, a multidrug pump inhibitor. Proc Natl Acad Sci U S A 2000; 97: 1433–7.
Rackova L, Majekova M, Kost'alova D, Stefek M . Antiradical and antioxidant activities of alkaloids isolated from Mahonia aquifolium. Structural aspects. Bioorg Med Chem 2004; 12: 4709–15.
Dai DZ . Vulnerable substrate and multiple ion channel disorder in a diseased heart will be new targets for antiarrhythmic therapy. Acta Pharmacol Sin 2000; 21: 289–95.
Zeng XH, Zeng XJ, Li YY . Efficacy and safety of berberine for congestive heart failure secondary to ischemic or idiopathic dilated cardiomyopathy. Am J Cardiol 2003; 92: 173–6.
Dai DZ, Dai Y . Induced ion currents and the endothelin pathway as targets for anti-arrhythmic agents. Curr Opin Investig Drugs 2008; 9: 1001–8.
Salehi S, Filtz TM . Berberine possesses muscarinic agonist-like properties in cultured rodent cardiomyocytes. Pharmacol Res 2011; 63: 335–40.
Zhang ZH, Deng AJ, Yu JQ, Li ZH, Wu LQ, Wang WJ, et al. Advance in studies on pharmacological activity of coptisine hydrochloride. Zhongguo Zhong Yao Za Zhi 2013; 38: 2750–4.
Cannillo M, Frea S, Fornengo C, Toso E, Mercurio G, Battista S, et al. Berberine behind the thriller of marked symptomatic bradycardia. World J Cardiol 2013; 5: 261–4.
Li MH, Zhang YJ, Yu YH, Yang SH, Iqbal J, Mi QY, et al. Berberine improves pressure overload-induced cardiac hypertrophy and dysfunction through enhanced autophagy. Eur J Pharmacol 2014; 728: 67–76.
Wang Z, Wu J, Zhou Q, Wang Y, Chen T . Berberine nanosuspension enhances hypoglycemic efficacy on streptozotocin induced diabetic C57BL/6 mice. Evid Based Complement Alternat Med 2015; 2015: 239749.
Lan J, Zhao Y, Dong F, Yan Z, Zheng W, Fan J, et al. Meta-analysis of the effect and safety of berberine in the treatment of type 2 diabetes mellitus, hyperlipemia and hypertension. J Ethnopharmacol 2015; 161: 69–81.
Yu L, Li F, Zhao G, Yang Y, Jin Z, Zhai M, et al. Protective effect of berberine against myocardial ischemia reperfusion injury: role of Notch1/Hes1-PTEN/Akt signaling. Apoptosis 2015; 20: 796–810.
Boengler K, Hilfiker-Kleiner D, Drexler H, Heusch G, Schulz R . The myocardial JAK/STAT pathway: from protection to failure. Pharmacol Ther 2008; 120: 172–85.
Bolli R, Dawn B, Xuan YT . Role of the JAK-STAT pathway in protection against myocardial ischemia/reperfusion injury. Trends Cardiovasc Med 2003; 13: 72–9.
Fuglesteg BN, Suleman N, Tiron C, Kanhema T, Lacerda L, Andreasen TV, et al. Signal transducer and activator of transcription 3 is involved in the cardioprotective signalling pathway activated by insulin therapy at reperfusion. Basic Res Cardiol 2008; 103: 444–53.
Bolli R, Dawn B, Xuan YT . Emerging role of the JAK-STAT pathway as a mechanism of protection against ischemia/reperfusion injury. J Mol Cell Cardiol 2001; 33: 1893–6.
Mascareno E, El-Shafei M, Maulik N, Sato M, Guo Y, Das DK, et al. JAK/STAT signaling is associated with cardiac dysfunction during ischemia and reperfusion. Circulation 2001; 104: 325–9.
Duan W, Yang Y, Yan J, Yu S, Liu J, Zhou J, et al. The effects of curcumin post-treatment against myocardial ischemia and reperfusion by activation of the JAK2/STAT3 signaling pathway. Basic Res Cardiol 2012; 107: 263.
Tsang CM, Cheung YC, Lui VW, Yip YL, Zhang G, Lin VW, et al. Berberine suppresses tumorigenicity and growth of nasopharyngeal carcinoma cells by inhibiting STAT3 activation induced by tumor associated fibroblasts. BMC Cancer 2013; 13: 619.
Yang N, Wang J, Liu C, Song Y, Zhang S, Zi J, et al. Berberine and limonin suppress IgE production by human B cells and peripheral blood mononuclear cells from food-allergic patients. Ann Allergy Asthma Immunol 2014; 113: 556–64.e4.
Chen K, Li G, Geng F, Zhang Z, Li J, Yang M, et al. Berberine reduces ischemia/reperfusion-induced myocardial apoptosis via activating AMPK and PI3K-Akt signaling in diabetic rats. Apoptosis 2014; 19: 946–57.
Davoodi-Semiromi A, Hassanzadeh A, Wasserfall CH, Droney A, Atkinson M . Tyrphostin AG490 agent modestly but significantly prevents onset of type 1 in NOD mouse; implication of immunologic and metabolic effects of a Jak-Stat pathway inhibitor. J Clin Immunol 2012; 32: 1038–47.
Kusaba M, Nakao K, Goto T, Nishimura D, Kawashimo H, Shibata H, et al. Abrogation of constitutive STAT3 activity sensitizes human hepatoma cells to TRAIL-mediated apoptosis. J Hepatol 2007; 47: 546–55.
Hartnett ME . The effects of oxygen stresses on the development of features of severe retinopathy of prematurity: knowledge from the 50/10 OIR model. Doc Ophthalmol 2010; 120: 25–39.
Ma LL, Zhang FJ, Qian LB, Kong FJ, Sun JF, Zhou C, et al. Hypercholesterolemia blocked sevoflurane-induced cardioprotection against ischemia-reperfusion injury by alteration of the MG53/RISK/GSK3beta signaling. Int J Cardiol 2013; 168: 3671–8.
Yu L, Sun Y, Cheng L, Jin Z, Yang Y, Zhai M, et al. Melatonin receptor-mediated protection against myocardial ischemia/reperfusion injury: role of SIRT1. J Pineal Res 2014; 57: 228–38.
Su H, Ji L, Xing W, Zhang W, Zhou H, Qian X, et al. Acute hyperglycaemia enhances oxidative stress and aggravates myocardial ischaemia/reperfusion injury: role of thioredoxin-interacting protein. J Cell Mol Med 2013; 17: 181–91.
Cordell GA, Quinn-Beattie ML, Farnsworth NR . The potential of alkaloids in drug discovery. Phytother Res 2001; 15: 183–205.
Vuddanda PR, Chakraborty S, Singh S . Berberine: a potential phytochemical with multispectrum therapeutic activities. Expert Opin Investig Drugs 2010; 19: 1297–307.
Tillhon M, Guaman Ortiz LM, Lombardi P, Scovassi AI . Berberine: new perspectives for old remedies. Biochem Pharmacol 2012; 84: 1260–7.
Xiao M, Men LN, Xu MG, Wang GB, Lv HT, Liu C . Berberine protects endothelial progenitor cell from damage of TNF-alpha via the PI3K/AKT/eNOS signaling pathway. Eur J Pharmacol 2014; 743: 11–6.
Zhang T, Yang S, Du J . Protective effects of berberine on isoproterenol-induced acute myocardial ischemia in rats through regulating HMGB1-TLR4 axis. Evid Based Complement Alternat Med 2014; 2014: 849783.
Toth A, Nickson P, Mandl A, Bannister ML, Toth K, Erhardt P . Endoplasmic reticulum stress as a novel therapeutic target in heart diseases. Cardiovasc Hematol Disord Drug Targets 2007; 7: 205–18.
Endo M, Oyadomari S, Suga M, Mori M, Gotoh T . The ER stress pathway involving CHOP is activated in the lungs of LPS-treated mice. J Biochem 2005; 138: 501–7.
Huang L, Xie H, Liu H . Endoplasmic reticulum stress, diabetes mellitus, and tissue injury. Curr Protein Pept Sci 2014; 15: 812–8.
Guo W, Jiang T, Lian C, Wang H, Zheng Q, Ma H . QKI deficiency promotes FoxO1 mediated nitrosative stress and endoplasmic reticulum stress contributing to increased vulnerability to ischemic injury in diabetic heart. J Mol Cell Cardiol 2014; 75: 131–40.
Wu XD, Zhang ZY, Sun S, Li YZ, Wang XR, Zhu XQ, et al. Hypoxic preconditioning protects microvascular endothelial cells against hypoxia/reoxygenation injury by attenuating endoplasmic reticulum stress. Apoptosis 2013; 18: 85–98.
Hao X, Yao A, Gong J, Zhu W, Li N, Li J . Berberine ameliorates pro-inflammatory cytokine-induced endoplasmic reticulum stress in human intestinal epithelial cells in vitro. Inflammation 2012; 35: 841–9.
Yu W, Sheng M, Xu R, Yu J, Cui K, Tong J, et al. Berberine protects human renal proximal tubular cells from hypoxia/reoxygenation injury via inhibiting endoplasmic reticulum and mitochondrial stress pathways. J Transl Med 2013; 11: 24.
Ferlito M, Wang Q, Fulton WB, Colombani PM, Marchionni L, Fox-Talbot K, et al. Hydrogen sulfide increases survival during sepsis: protective effect of CHOP inhibition. J Immunol 2014; 192: 1806–14.
Aringer M, Cheng A, Nelson JW, Chen M, Sudarshan C, Zhou YJ, et al. Janus kinases and their role in growth and disease. Life Sci 1999; 64: 2173–86.
Grote K, Luchtefeld M, Schieffer B . JANUS under stress--role of JAK/STAT signaling pathway in vascular diseases. Vascul Pharmacol 2005; 43: 357–63.
Horvath CM . STAT proteins and transcriptional responses to extracellular signals. Trends Biochem Sci 2000; 25: 496–502.
Kurdi M, Booz GW . Can the protective actions of JAK-STAT in the heart be exploited therapeutically? Parsing the regulation of interleukin-6-type cytokine signaling. J Cardiovasc Pharmacol 2007; 50: 126–41.
Hattori R, Maulik N, Otani H, Zhu L, Cordis G, Engelman RM, et al. Role of STAT3 in ischemic preconditioning. J Mol Cell Cardiol 2001; 33: 1929–36.
Tian Y, Zhang W, Xia D, Modi P, Liang D, Wei M . Postconditioning inhibits myocardial apoptosis during prolonged reperfusion via a JAK2-STAT3-Bcl-2 pathway. J Biomed Sci 2011; 18: 53.
Luo X, Gu J, Zhu R, Feng M, Zhu X, Li Y, et al. Integrative analysis of differential miRNA and functional study of miR-21 by seed-targeting inhibition in multiple myeloma cells in response to berberine. BMC Syst Biol 2014; 8: 82.
Bao M, Cao Z, Yu D, Fu S, Zhang G, Yang P, et al. Columbamine suppresses the proliferation and neovascularization of metastatic osteosarcoma U2OS cells with low cytotoxicity. Toxicol Lett 2012; 215: 174–80.
Muralimanoharan SB, Kunnumakkara AB, Shylesh B, Kulkarni KH, Haiyan X, Ming H, et al. Butanol fraction containing berberine or related compound from nexrutine inhibits NFkappaB signaling and induces apoptosis in prostate cancer cells. Prostate 2009; 69: 494–504.
Li Y, Zhu W, Tao J, Xin P, Liu M, Li J, et al. Fasudil protects the heart against ischemia-reperfusion injury by attenuating endoplasmic reticulum stress and modulating SERCA activity: the differential role for PI3K/Akt and JAK2/STAT3 signaling pathways. PLoS One 2012; 7: e48115.
Negoro S, Kunisada K, Fujio Y, Funamoto M, Darville MI, Eizirik DL, et al. Activation of signal transducer and activator of transcription 3 protects cardiomyocytes from hypoxia/reoxygenation-induced oxidative stress through the upregulation of manganese superoxide dismutase. Circulation 2001; 104: 979–81.
Yang Y, Duan W, Jin Z, Yi W, Yan J, Zhang S, et al. JAK2/STAT3 activation by melatonin attenuates the mitochondrial oxidative damage induced by myocardial ischemia/reperfusion injury. J Pineal Res 2013; 55: 275–86.
Kurdi M, Sivakumaran V, Duhe RJ, Aon MA, Paolocci N, Booz GW . Depletion of cellular glutathione modulates LIF-induced JAK1-STAT3 signaling in cardiac myocytes. Int J Biochem Cell Biol 2012; 44: 2106–15.
Zgheib C, Kurdi M, Zouein FA, Gunter BW, Stanley BA, Zgheib J, et al. Acyloxy nitroso compounds inhibit LIF signaling in endothelial cells and cardiac myocytes: evidence that STAT3 signaling is redox-sensitive. PLoS One 2012; 7: e43313.
Zhang Y, Li X, Zou D, Liu W, Yang J, Zhu N, et al. Treatment of type 2 diabetes and dyslipidemia with the natural plant alkaloid berberine. J Clin Endocrinol Metab 2008; 93: 2559–65.
Kong WJ, Wei J, Zuo ZY, Wang YM, Song DQ, You XF, et al. Combination of simvastatin with berberine improves the lipid-lowering efficacy. Metabolism 2008; 57: 1029–37.
Acknowledgements
This study was supported by grants from the International Scientific and Technological Cooperation and Exchange Project of Shaanxi Province (No 2015KW-047), the Social Development Project of Shaanxi Province (No 2015SF104 and 2012K15-02-01), the National Natural Science Foundation of China (No 81470415, 81270170, 81200151, and 81470411), the National 12th Five Year Plan for Science and Technology Support (No 2011BAI11B20), the Science and Technology Co-ordinating Innovative Engineering Project of Shaanxi Province (No 2013KTCL03-01), the Natural Science Foundation of Shaanxi Province (No 2014JM4106), and the Subject-Boosting Project of Xijing Hospital (No XJZT14203, XJGX12C11, and XJGX13LC15).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Zhao, Gl., Yu, Lm., Gao, Wl. et al. Berberine protects rat heart from ischemia/reperfusion injury via activating JAK2/STAT3 signaling and attenuating endoplasmic reticulum stress. Acta Pharmacol Sin 37, 354–367 (2016). https://doi.org/10.1038/aps.2015.136
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2015.136
Keywords
This article is cited by
-
Endoplasmic reticulum stress and unfolded protein response in cardiovascular diseases
Nature Reviews Cardiology (2021)
-
Unfolded protein response during cardiovascular disorders: a tilt towards pro-survival and cellular homeostasis
Molecular and Cellular Biochemistry (2021)
-
Identification of berberine as a novel drug for the treatment of multiple myeloma via targeting UHRF1
BMC Biology (2020)
-
Berberine Attenuates Cardiac Hypertrophy Through Inhibition of mTOR Signaling Pathway
Cardiovascular Drugs and Therapy (2020)
-
Berberine Ameliorates Oxygen-glucose Deprivation/Reperfusion-induced Apoptosis by Inhibiting Endoplasmic Reticulum Stress and Autophagy in PC12 Cells
Current Medical Science (2020)
