
Abstract
Aim:
To investigate the role of extracellular signal-regulated kinases (ERKs) in sevoflurane post-conditioning induced cardioprotection in vitro.
Methods:
Isolated rat hearts were subjected to 30 min ischemia followed by 120 min reperfusion (I/R). Sevoflurane post-conditioning was carried out by administration of O2-enriched gas mixture with 3% sevoflurane (SEVO) for 15 min from the onset of reperfusion. Cardiac functions, myocardial infarct size, myocardial ATP and NAD+ contents, mitochondrial ultrastructure, and anti-apototic and anti-oncosis protein levels were measured.
Results:
Sevoflurane post-conditioning significantly improved the heart function, decreased infarct size and mitochondria damage, and increased myocardial ATP and NAD+ content in the I/R hearts. Furthermore, sevoflurane post-conditioning significantly increased the levels of p-ERK and p-p70S6K, decreased the levels of porimin, caspase-8, cleaved caspase-3, and cytosolic cytochrome c in the I/R hearts. Co-administration of the ERK1/2 inhibitor PD98059 (20 μmol/L) abolished the sevoflurane-induced protective effects against myocardial I/R.
Conclusion:
Sevoflurane post-conditioning protects isolated rat hearts against myocardial I/R injury and inhibits cell oncosis and apoptosis via activation of the ERK1/2 pathway.
Similar content being viewed by others


Introduction
The prevalence of ischemic heart diseases (IHDs) is continuously increasing around the world1,2. Patients suffering from these diseases are often at increased risk of myocardial I/R injury, which is an important reason for increasing perioperative complications and mortality3. A series of brief episodes of ischemia and reperfusion at the onset of reperfusion can reduce myocardial infarct size, known as “ischemic post-conditioning”4,5. However, it is difficult to apply ischemic post-conditioning to clinical practice because well-controlled, short episodes of myocardial ischemia are almost impossible to perform. Sevoflurane is a volatile anesthetic and has been widely used in clinical practice. Anesthetic post-conditioning was first reported to protect myocardium from I/R injury in 20066. In recent years, many studies, including experimental investigations7,8 and clinical practice9,10,11 have demonstrated that sevoflurane post-conditioning protects the hearts against myocardial I/R injury. Although many studies have addressed anesthetic post-conditioning in recent years, the underlying mechanisms have not been fully elucidated.
Extracellular-signal regulated kinases (ERKs), members of the subfamilies of mitogen-activated protein kinases (MAPKs), are well known to modulate cellular transcriptional activities, cell proliferation, differentiation, survival and apoptosis12. ERK1/2 pathway has previously been implicated in ischemic and anesthetic post-conditioning13,14 and plays a critical role in myocardial protection against I/R injury. Phosphorylation of ERK up-regulates expression of its downstream target p-p70S6K. ERK1/2 pathway, activated by sevoflurane post-conditioning, has been shown to regulate mitochondrial permeability transition thereby inhibiting caspase activation. It is believed that the mitochondrial permeability transition pore (mPTP), which is a crucial end-effector in myocardium I/R injury, opens in the first few minutes after reperfusion. Increased mPTP opening is a major cause of cardiomyocyte apoptosis and oncosis following myocardium I/R injury15,16.
Apoptosis is a process of programmed cell death process, and oncosis is an important non-apoptotic mode. ATP levels determine which pathway is taken, apoptosis, with normal intracellular ATP levels or oncosis, with intracellular ATP deficiency17. Caspase-8 plays an important role in caspase activation cascades, activating downstream effector caspases such as caspase-3, -6, and -7 that lead to apoptosis18. On the other hand, oncotic cell death can be mediated by porimin, a highly glycosylated protein and member of the cell membrane-associated mucin family located on the cell surface19. For example, oncosis-like cell death can be mediated by introduction of porimin cDNA into Jurkat cells20.
The mechanisms underlying the cardioprotective actions of sevoflurane post-conditioning remain unclear. Here we test the hypothesis that activation of the ERK1/2 pathway is necessary for sevoflurane post-conditioning and involves reducing oncosis protein porimin expression and inhibiting mPTP opening in the isolated perfused rat hearts subjected to global ischemia.
Materials and methods
Adult, healthy male Sprague-Dawley rats, weighing 280–330 g, and of healthy grade were maintained from Soochow University School of Medicine Experimental Animal Center. The animal experiments were approved by the Committee of the Medical College of Soochow University (Suzhou, China; License Number: 2002-0008 Grade II).
Experimental protocols
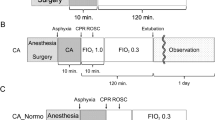
One hundred and twenty-six isolated perfused rat hearts were randomly divided into 6 groups: control group (CON), ischemia/reperfusion group (I/R), sevoflurane postconditioning group (SEVO), DMSO group (DMSO), PD98059 group (PD), and PD98059+SEVO group (PD+SEVO). Except the CON group, each rat was subjected to 30 min of global ischemia followed by 2 h of reperfusion. SEVO group received 2.5% (v/v) sevoflurane for 15 min at onset of reperfusion, then reperfusion the hearts for 105 min. PD group received the ERK1/2 specific inhibitor PD98059 (20 μmol/L) (Sigma-Aldrich, St Louis, MO, USA) for 15 min at onset of reperfusion. The experimental design is shown in Figure 1.

Schematic diagram depicting the experimental protocol (n=18 per group). With the exception of the CON group, each rat heat was subjected to 30 min of global ischemia followed by 2 h of reperfusion and was assigned to the I/R group. Sevoflurane (3.0%) was administered for 15 min at the onset of reperfusion in the SEVO group. The PD98059 vehicle DMSO (0.2%) was administered for 15 min at the onset of reperfusion in the DMSO group. The selective ERK1/2 inhibitor PD98059 (20 μmol/L) was administered for 15 min at the onset of reperfusion in the PD group. Both PD98059 in DMSO and 3% sevoflurane were administered for 15 min at the onset of reperfusion in the PD+SEVO group. In a parallel set of hearts (n=6), the experiments were terminated after 15 min of reperfusion to obtain samples for NAD+ measurements. CON, control; I/R, ischemia/reperfusion; SEVO, sevoflurane post-conditioning; DMSO, dimethyl sulfoxide; PD, PD98059; ISC, ischemia.
Langendorff isolated heart perfusion model
Krebs-Henseleit (K-H) solution configuration (mmol/L): NaCl 118.0, KCl 4.8, KH2PSO4 1.2, NaHCO3 25.0, MgSO4 1.2, CaCl2 2.5, glucose 11.0, and pH 7.35–7.45. After pre-access 95% O2–5% CO2 for 30 min, K-H solution was maintained at 37 °C by an external circulation (Alcott Biotechnology Co, Ltd, Shanghai, China). Rats were anaesthetized with sodium pentobarbital 50 mg/kg and heparinized by intraperitoneal injection, then the hearts were quickly excised and mounted on a Langendorff apparatus via the aorta for retrograde perfusion with K-H solution at constant pressure (10 kPa)21. The pump was adjusted to maintain coronary transudes to 12 mL/min.
Cardiac functions
Ventricular chamber pressures were taken and maintained with a cuff pressure transducer (SIA Industrial & Trade, Beijing, China). A saline-filled latex balloon was inserted into the left ventricle through the mitral valve to monitor the heart function14. The cuff volume was adjusted to achieve a stable left ventricular end-diastolic pressure (LVEDP) of 5–10 mmHg during initial equilibration. Cardiac functions, including left ventricular systolic pressure (LVSP), left ventricular end-diastolic pressure (LVEDP), ±dp/dtmax and heart rate (HR) were recorded at 30 min of equilibration (T0), 30 min (T1), 60 min (T2), 90 min (T3), and 120 min (T4) after reperfusion by Med Lab 6.0 software. Rats with refractory ventricular fibrillation, frequent arrhythmia, LVSP<75 mmHg or HR<180 beats/min were excluded.
Infarct size determination
Myocardial infarct size was determined as previously described22. Briefly, at the end of reperfusion, the rat hearts were removed and cut into 6 pieces in cross-section. The hearts were stained with 1% 2,3,5-triphenyltetrazolium chloride triazole (TTC, Sigma-Aldrich, St Louis, MO, USA) in pH 7.4 buffer at 37 °C for 20 min and fixed overnight in 10% formalin. Because TTC stains viable tissue a deep red color, unstained tissue was presumed to be infarcted. The infarct size was calculated for each slice, and reported as the percent of infarct area divided by the total area at risk by Alpha Ease FC Imaging System.
Transmission electron microscopy
Myocardial ultrastructural alterations were detected by transmission electron microscopy (TEM)23. Rat hearts were rapidly removed at the end of reperfusion and a small piece (1 mm3) of tissue was dissected from left ventricular and fixed in 2.5% glutaraldehyde in 0.1 mol/L sodium cacodylate buffer (pH 7.4) for 4 h. Tissues were post-fixed in 1% osmium tetroxide in 1% K4Fe(CN)6, dehydrated through graded concentrations of ethanol and propylene oxide, embedded in Epon812 and then sectioned using an ultramicrotome. Longitudinal sections were placed onto copper grids which were stained with uranyl acetate and lead nitrate, and visualized on an H-600 electron microscope (Hitachi Limited, Tokyo, Japan). Three fields were examined for each sample at ×8000 magnification.
Myocardial ATP content measurement
Myocardial ATP concentration was determined by using a bioluminescence method as described24,25. A luminometric method based on the luciferin-luciferase reaction was used to quantify ATP in myocardium by using an ATP assay kit (Beyotime Institute of Biotechnology, Haimen, China). The concentration of phosphocreatine in myocardium was determined by using the reverse phase-high performance liquid chromatography (RP-HPLC) methods described26. The phosphocreatine standard was from Sigma-Aldrich. The concentration of glycogen in myocardium was determined with a chemistry colorimetry method by using a glycogen detection kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China).
Myocardial NAD+ content measurement
The left ventricular tissues were taken after 15 min of reperfusion for measuring nicotinamide adenine dinucleotide (NAD+) levels. NAD+ was extracted from left ventricular tissues at risk using perchloric acid27. Freeze-clamped tissue (30 mg) was powdered in a mortar and completely mixed with 150 mL 0.6 mol/L perchloric acid. Subsequently, the mixture was homogenized, neutralized with 3 mol/L KOH and centrifuged for 10 min. The centrifuged sediment was dissolved in 0.1 mol/L NaOH solution. The dilutions of the supernatant samples were removed and NAD+ concentrations were determined fluorometrically using alcohol dehydrogenase (Sigma-Aldrich, St Louis, MO, USA) at a wavelength of 460 nm in a Multi-frequency Phase ISS K2 Spectrofluorimeter (ILC Technology Inc, Sunnyvale, CA, USA).
Mitochondrial and cytoplasm preparation and protein extraction
The left ventricular tissue was taken at the end of reperfusion and homogenized in ice-cold sucrose buffer, which contained 300 mmol/L sucrose, 5 mmol/L TES, 0.2 mmol/L EGTA. After the homogenate was centrifuged at 800×g for 10 min, the supernatant was further centrifuged at 10 000×g for 15 min. The resulting pellet contained mitochondria. The supernatant after 10 000×g centrifugation was centrifuged at 100 000×g for 60 min and this supernatant contained the cytoplasm fraction.
Total or mitochondrial protein extracts were obtained by homogenizing heart tissue samples or mitochondrial pellets in lysis buffer [20 mmol/L Tris-HCl (pH 7.4), 150 mmol/L NaCl, 1 mmol/L EDTA, 1 mmol/L NaVO3, 1 mmol/L NaF, 2.5 mmol/L Na4P2O7, 1% NP40, 0.1% SDS, 1 mmol/L DTT, 1 mmol/L PMSF]. The pellet was washed and resuspended in lysis buffer, and a complete proteinase inhibitor cocktail (Sigma, one tablet per 10 mL). The homogenates were vortexed under ultrasound and then centrifuged at 14 000×g for 10 min at 4 °C. BCA protein assay kit (Beyotime Institute of Biotechnology, Haimen, China) was used for determining protein concentrations in each fraction and the lysate was transferred to the same concentration.
Western blot analyses of proteins
Equivalent amounts (30 μg each) of protein from cytosol and mitochondria were thoroughly mixed with 2×laemmeli buffer and then heated at 97.0°C for 5 min. Subsequently, denatured protein extracts were electrophoresed on 10% or 15% SDS-PAGE gels and then transferred to a polyvinylidine fluoride (PVDF) membrane. After blocking with 5% non-fat milk for 2 h, PVDF membranes were incubated with following primary antibodies at 4oC overnight: anti-ERK1/2 (1:1000, Cell Signaling Technology, Danvers, MA, USA), anti-p-ERK1/2 (1:1000, Cell Signaling Technology), anti-p70S6K (1:1000, Cell Signaling Technology), anti-p-p70S6K (1:1000, Cell Signaling Technology), anti-Porimin (1:1000, Sigma-Aldrich), anti- cytochrome c (1:1000, Santa Cruz, Dallas, TX, USA), anti-caspase-8 (1:1000, Santa Cruz) and anti-cleaved caspase-3 (1:1000, Santa Cruz). The primary antibody binding was detected with a secondary anti-mouse or rabbit antibody (1:5000) and visualized with enhanced chemiluminescence. Anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH, 1:2000, AG019, Beyotime Institute of Biotechnology) and anti-prohibitin (PHB) (1:1000) (Cell Signaling Technology) were used as internal control for cytoplasm and mitochondria, respectively. Semiquantitative analysis of the optical densities of protein bands was performed using Image J software. Band densities obtained from p-ERK1/2 and p-p70S6K were normalized against the concentrations of t-ERK1/2 and t-p70S6K in the same samples.
Statistical analysis
Graphpad Prism 4.00 statistical software were used for statistical processing. Measurement data were expressed as mean±standard deviation (mean±SD). Data were analyzed by using one-way analysis of variance followed by Tukey multiple-comparison post test, P<0.05 was considered statistically significant.
Results
Cardiac functions results
Baseline hemodynamics among all groups were similar (P>0.05). Compared with equilibration, in addition to the CON group, LVSP and HR, ±dp/dtmax obviously decreased, LVEDP significantly increased during reperfusion among the experimental groups (P<0.05). LVSP, HR, and ±dp/dtmax significantly decreased and LVEDP significantly increased in the other groups compared with the CON group at 30, 60, 90, and 120 min of reperfusion (P<0.05). LVSP, HR, and ±dp/dtmax increased, LVEDP significantly decreased in the SEVO group, relative to the I/R group (P<0.05). The DMSO, PD, and PD+SEVO groups were similar to the I/R group (P>0.05) (Table 1).
Myocardial infarct size
Myocardial infarct size in the SEVO group (23.44%±1.98%) was decreased compared with the I/R group (51.97%±1.63%) (P<0.05); however, this was abolished by using PD98059 at PD+SEVO group (49.32%±1.82%). Infarct size in the DMSO, PD, and SEVO+PD groups were 49.60%±1.40%, 49.70%±1.39%, and 49.32%±1.82%, respectively, with no statistical differences compared to the I/R group (P>0.05) (Figure 2).

Myocardial infarct sizes of all groups expressed as the percentage of infarct size normalized to the area at risk of isolated rat hearts after 2 h of reperfusion. Infarct size was determined using 1% triphenyltetrazolium chloride staining. The size of the infarcted myocardium was calculated as an area ratio (pale area/total area of the myocardium at risk). Data are presented as mean±SD. n=6. bP<0.05, versus CON. CON, control; I/R, ischemia/reperfusion; SEVO, sevoflurane post-conditioning; DMSO, dimethyl sulfoxide; PD, PD98059.
Morphology of mitochondrion with transmission electron microscopy analysis
Using transmission electron microscopy (TEM), we found that mitochondrial volume density was similar within each groups. However, left ventricular myocytes exhibit significant morphological defects and oncosis, including altered cristae density and architecture in the other groups, relative to the CON group. Most mitochondria exhibited typical morphology in the CON group. The SEVO group had less mitochondria damage, swelling of the matrix and vacuolization of cristae. Magnification was set at ×8000 (Figure 3).

Myocardial mitochondrial ultrastructure. Transmission electron microscopy of myocardium after reperfusion in all experimental groups. These are representative images taken of mounted sections per heart. Myocardial cells exhibit significant swelling of mitochondria with disruption of cristae in the I/R, DMSO, PD, PD+DMSO groups. The SEVO group had less mitochondria damage. Magnification was set at ×8000. n=3. CON, control; I/R, ischemia/reperfusion; SEVO, sevoflurane post-conditioning; DMSO, dimethyl sulfoxide; PD, PD98059.
Myocardial ATP content
The ATP content in the CON group was significantly greater than that of the other groups (P<0.05). The ATP loss was reduced in the SEVO group compared to the I/R group (P<0.05). There were no statistical differences among the I/R, DMSO, PD, and PD+SEVO groups (P>0.05) (Figure 4).

Myocardial ATP content after reperfusion. Compared with the CON group, the ATP content in the other groups significantly decreased. Compared with the I/R group, the ATP content in the SEVO group significantly increased. There were no obvious differences between the other groups. Data are presented as mean±SD. n=6. bP<0.05, versus CON. eP<0.05, versus I/R group. CON, control; I/R, ischemia/reperfusion; SEVO, sevoflurane post-conditioning; DMSO, dimethyl sulfoxide; PD, PD98059.
Myocardial NAD+ content
The NAD+ content in the other groups was much lower than the CON group (P<0.05). However, NAD+ loss was reduced in the SEVO group. There were no significant differences among the other groups: I/R, DMSO, PD, and PD+SEVO (P>0.05) (Figure 5).

Myocardial NAD+ content after 15 min of reperfusion. Compared with the CON group, the NAD+ content in the other groups decreased. Compared with the I/R group, the NAD+ content in the SEVO group significantly increased. There were no obvious differences between the other groups. Data are presented as mean±SD. n=6. bP<0.05, versus CON. eP<0.05, versus I/R group. CON, control; I/R, ischemia/reperfusion; SEVO, sevoflurane post-conditioning; DMSO, dimethyl sulfoxide; PD, PD98059; NAD, nicotinamide adenine dinucleotide.
Western blot
Compared with the CON group, the levels of p-ERK1/2 and levels of p-p70S6K at the end of reperfusion in the I/R group were increased (P<0.05). Compared with the I/R group, the ratios of p-ERK1/2/total and levels of p-p70S6K/total in the SEVO group were increased further (P<0.05); whereas, the ratios of p-ERK1/2/total and p-p70S6K/total in the I/R and the DMSO groups were similar (P>0.05) (Figure 6).

Western blot of p-ERK, t-ERK, p-p70S6K, and t-p70S6K from left ventricular samples acquired after the end of reperfusion. p-ERK/t-ERK and p-p70S6K/t-p70S6K ratios significantly increased in the SEVO group. PD98059 administration upon reperfusion abolished increased levels of p-ERK1/2 and p-p70S6K induced by sevoflurane. Data are presented as mean±SD. n=3. bP<0.05, versus CON. eP<0.05, versus I/R group. CON, control; I/R, ischemia/reperfusion; SEVO, sevoflurane post-conditioning; DMSO, dimethyl sulfoxide; PD, PD98059.
The levels of porimin, caspase-8 and cleaved caspase-3 in the other groups were significantly greater than that of the CON group (P<0.05). The expression of porimin, caspase-8 and cleaved caspase-3 in the SEVO group was reduced compared to the other I/R groups (P<0.05). There were no significant differences among the other groups: I/R, DMSO, PD, and PD+SEVO (P>0.05) (Figure 7 and 8).

Western blot of porimin from left ventricular samples were acquired at the end of reperfusion. Compared with the CON group, the expression of porimin in the other groups significantly increased. Compared with the I/R groups, the expression of porimin in the SEVO group decreased. There were no obvious differences between levels in the I/R, DMSO, PD, PD+SEVO groups. Data are presented as mean±SD. n=3. bP<0.05, versus CON. eP<0.05, versus I/R group. CON, control; I/R, ischemia/reperfusion; SEVO, sevoflurane post-conditioning; DMSO, dimethyl sulfoxide; PD, PD98059.

Western blot of cleaved caspase-3 (A) and caspase-8 (B) from left ventricular samples were acquired at the end of reperfusion. Compared with the CON group, the expression of cleaved caspase-3 and caspase-8 in the other groups significantly increased. Compared with the I/R groups, the expression of cleaved caspase-3 and caspase-8 in the SEVO group decreased. There were no obvious differences between levels in the I/R, DMSO, PD, PD+SEVO groups. Data are presented as mean±SD. n=3. bP<0.05, versus CON. eP<0.05, versus I/R group. CON, control; I/R, ischemia/reperfusion; SEVO, sevoflurane post-conditioning; DMSO, dimethyl sulfoxide; PD, PD98059.
Compared with the CON group, the levels of Cyt c significantly increased in cytosol compared to the other groups (P<0.05) and dramatically decreased in mitochondria (P<0.05). The expression of Cyt c in the SEVO group clearly decreased in cytosol (P<0.05) and significantly increased in mitochondria, relative to the other I/R groups (P<0.05). The levels of Cyt c in cytosol and mitochondria in the other groups: I/R, DMSO, PD, and PD+SEVO groups were similar (P>0.05) (Figure 9A and 9B).

Western blot of Cyt c in cytosol (A) and mitochondria (B) from left ventricular samples were acquired at the end of reperfusion. Compared with the CON group, the expression of Cyt c in the other groups significantly increased in cytosol and obviously decreased in mitochondria (P<0.05). Compared with the I/R group, the expression of Cyt c in the SEVO group decreased in cytosol and significantly increased in mitochondria (P<0.05). There were no statistical differences among the other groups: I/R, DMSO, PD, and PD+SEVO groups (P>0.05). Data are presented as mean±SD. n=3. bP<0.05, versus CON. eP<0.05, versus I/R group. CON, control; I/R, ischemia/reperfusion; SEVO, sevoflurane post-conditioning; DMSO, dimethyl sulfoxide; PD, PD98059; Cyt c, Cytochrome c.
Discussion
Recently, many other researchers have demonstrated that anesthetic post-conditioning is mediated through several signaling elements, including mitochondrial ATP-sensitive potassium channels (mito-KATP), mitochondrial permeability transition pore (mPTP), phosphatidylinositol-3-kinase (PI3K)-ERK1/2 signal pathway14. In the present study, we chose 3% sevoflurane to post-condition the isolated rat hearts in a Langendorff perfusion system28. Intracellular mitogen-activated protein kinases (MAPKs) signaling cascades play a key role in the pathogenesis of cardiac and vascular disease. ERK1/2, a member of the MAPKs, is concerned with the regulation of cell proliferation, differentiation, survival and apoptosis. Although most researchers have maintained that the phosphorylation of ERK1/2 played an important role in protecting against myocardial I/R injury, others have shown that ERK1/2 phosphorylation may aggravate myocardial cell injury29. ERK phosphorylation not only up-regulates its downstream target p-p70S6K and down-regulates the oncosis and apoptosis related protein expression, but also reduces the release of Cyt c from mitochondria into the cytosol and inhibits mPTP opening.
Sevoflurane post-conditioning was abolished by using 20 μmol/L PD98059, a specific inhibitor of ERK. It suggests that activation of the ERK1/2 pathway is required for the myocardial protective effects of sevoflurane post-conditioning. We found sevoflurane increased p-ERK1/2 and p-p70S6K; whereas, PD98059 completely inhibited the activation of ERK1/2 and partially inhibited the activation of p70S6K. p-p70S6K plays a role in reducing cell apoptosis and regulating the activity of pro-apoptotic protein BAD to protect the hearts30.
In addition to their role in cellular energy metabolism, mitochondria are now recognized as central players in cell death. By using TEM, we found myocardial mitochondrial injury, such as cristae swelling and vacuolization in the I/R group were more prominent than in the SEVO group which indicated that sevoflurane post-conditioning reduced myocardial oncosis. Myocardial NAD+ content is inversely proportional to mPTP opening31, we found that sevoflurane post-conditioning reduced NAD+ release. This indicated that sevoflurane post-conditioning protects isolated rat hearts against I/R injury by inhibiting mPTP opening. ATP is the main source of cellular energy metabolism, and functional recovery after myocardial I/R puts further demands on energy supply. As expected, ATP content in the SEVO group was significant higher than the other injury groups. Thus, we can conclude that sevoflurane post-conditioning protects cardiomyocytes by improving myocardial energy metabolism after ischemia and during reperfusion injury, thus inhibiting apoptosis. In addition, the release of Cyt c from mitochondria into the cytosol can trigger the activity of caspases, which is a key step in apoptosis32.
By Western blot, we found that sevoflurane post-conditioning reduced cytosolic Cyt c, which confirmed that sevoflurane post-conditioning could reduce the caspase cascade reaction. Therefore, it is a strong possibility that ERK1/2 pathway is required for sevoflurane post-conditioning not only by suppressing apoptotic cascades, but also reducing oncosis signaling.
Porimin is a highly glycosylated protein that can be classified as a member of the cell membrane-associated mucin family and serves as a novel cell surface receptor mediating oncotic cell death20. Porimin is activated by various stressors, such as hypoxia, the lack of metabolic substrates, or rapid ATP depletion33,34. When activated it quickly combines with its ligand anti-porimin mAb, resulting in membrane structural damage, accelerating oncosis20. Oncosis is an important non-apoptotic type of cell death. Cell oncosis leads to necrosis with karyolysis, characterized by cellular swelling, membrane blebbing and increased membrane permeability, and stands in contrast to apoptosis, which leads to necrosis with karyorhexis and cell shrinkage34. Oncosis and apoptosis represent two different cell death pathways, but they could be produced by the same stimuli. It has been demonstrated that oncosis is a major contributor to ischemic cardiomyocyte death35,36. Oncosis also occurs in myocardial I/R injury, and porimin plays an important role. By Western blot, we found that I/R inury increased porimin levels whereas sevoflurane reduced this increase, and this reduction was reversed by PD98059. Thus, sevoflurane post-conditioning acting through the ERK1/2 pathway inhibits porimin expression, and thereby decreases cell oncosis, as well as apoptosis.
In our study, several limitations have to be mentioned. We only examined one inhaled anesthetic, sevoflurane. Whether other volatile anesthetics protect isolated rat hearts against myocardial I/R injury through this same post-conditioning mechanism needs further study. Although the experiment strongly supports the idea that ERK1/2 pathway is necessary for sevoflurane post-conditioning, further experimentation is needed to define the specific mechanisms that underlie its cardioprotective effect. While we chose a relatively low concentration of ERK1/2 inhibitor, we cannot eliminate the possibility that other protein kinase activities may have been affected.
In summary, our findings showed that sevoflurane post-conditioning protects isolated rat hearts against myocardial I/R injury. ERK1/2/p70S6K pathway plays an important role in sevoflurane post-conditioning against myocardial I/R injury in isolated rat hearts by inhibiting the activation of oncosis and apoptosis proteins, reducing the release of Cyt c from mitochondria into the cytosol, inhibiting mPTP opening and improving myocardial energy metabolism. The ERK1/2 pathway appears to modulate not only anti-apoptotic pathways, but also anti-oncosis pathways in the isolated rat heart I/R model. Taken as a whole, our present findings provide mechanistic insights into the cardioprotection of sevoflurane post-conditioning and underscore its protection in oncosis and apoptosis where could be part of a therapeutic strategy to treat peri-operative ischemic events.
Author contribution
Chen WANG designed the research and provided financial support; Hong XIE collected and analyzed data, detected the protein expression; Li-xin LIU and Mario REBECCHI helped design the study and compose the manuscript; Jiang ZHU modified the paper and guided scientific research; Jing ZHANG conducted studies of the morphology of mitochondria, and measured the myocardial ATP and myocardial NAD+ content; Su-mei HU took part in establishing the isolated rat heart model.
References
Pepine CJ, Nichols WW . The pathophysiology of chronic ischemic heart disease. Clin Cardiol 2007; 30: 4–9.
Xu K, George I, Klotz S, Hay I, Xydas S, Zhang G, et al. Erythropoietin derivate improves left ventricular systolic performance and attenuates left ventricular remodeling in rats with myocardial infarct-induced heart failure. J Cardiovasc Pharmacol 2010; 56: 506–12.
Kertai MD, Klein J, Bax JJ, Poldermans D . Predicting perioperative cardiac risk. Prog Cardiovasc Dis 2005; 47: 240–57.
Zhao ZQ . Postconditioning in reperfusion injury: a status report. Cardiovasc Drugs Ther 2010; 24: 265–79.
Zhao ZQ, Corvera JS, Halkos ME, Kerendi F, Wang NP, Guyton RA, et al. Inhibition of myocardial injury by ischemic post-conditioning during reperfusion: Comparison with ischemic preconditioning. Am J Physiol Heart Circ Physiol 2003; 285: 579–88.
Zhao ZQ, Vinten JJ . Postconditioning: reduction of reperfusion- induced injury. Cardiovasc Res 2006; 70: 200–11.
Yao YT, Fang NX, Shi CX, Li LH . Sevoflurane and clinical practices postconditioning protects isolated rat hearts against ischemia-reperfusion injury. Chin Med J 2010; 123: 1320–8.
Fang NX, Yao YT, Shi CX, Li LH . Attenuation of ischemia-reperfusion injury by sevoflurane postconditioning involves protein kinase B and glycogen synthase kinase 3 beta activation in isolated rat hearts. Mol Biol Rep 2010; 37: 3763–9.
De Hert S, Vlasselaers D, Barbé R, Ory JP, Dekegel D, Donnadonni R, et al. Comparison of volatile and non volatile agents for cardioprotection during on-pump coronary surgery. Anaesthesia 2009; 64: 953–60.
Piriou V, Mantz J, Goldfarb G, Kitakaze M, Chiari P, Paquin S, et al. Sevoflurane preconditioning at 1 MAC only provides limited protection in patients undergoing coronary artery bypass surgery: a randomized bi-centre trial. Br J Anaesth 2007; 99: 624–31.
Nader ND, Li CM, Khadra WZ, Reedy R, Panos AL . Anesthetic myocardial protection with sevoflurane. J Cardiothorac Vasc Anesth 2004; 18: 269–74.
Boulton TG, Yancopoulos GD, Gregory JS, Slaughter C, Moomaw C, Hsu J, et al. An insulin-stimulated protein kinase similar to yeast kinases involved in cell cycle control. Science 1990; 249: 64–7.
Inamura Y, Miyamae M, Sugioka S, Domae N, Kotani J . Sevoflurane postconditioning prevents activation of Caspase-3 and 9 through antiapoptotic signaling after myocardial ischemia-reperfusion. J Anesth 2010; 24: 215–24.
Chen HT, Yang CX, Li H, Zhang CJ, Wen XJ, Zhou J, et al. Cardioprotection of sevoflurane postconditioning by activating extracellular signal-regulated kinase 1/2 in isolated rat hearts. Acta Pharmacol Sin 2008; 29: 931–41.
Stanley WC, Hoppel CL . Mitochondrial dysfunction in heart failure: potential for therapeutic interventions? Cardiovasc Res 2000; 45: 805–6.
Lesnefsky EJ, Moghaddas S, Tandler B, Kerner J, Hoppel CL . Mitochondrial dysfunction in cardiac disease: ischemia-reperfusion, aging, and heart failure. J Mol Cell Cardiol 2001; 33: 1065–89.
Chang SH, Phelps PC, Berezesky IK, Ebersberger ML Jr, Trump BF . Studies on the mechanisms and kinetics of apoptosis induced by microinjection of cytochrome in rat kidney tubule epithelial cells (NRK252E). Am J Pathol 2000; 156: 637–49.
Guo XX, Guo Q, Li Y, Lee SK, Wei XN, Jin YH . Ginsenoside Rh2 induces human hepatoma cell apoptosisvia bax/bak triggered cytochrome c release and caspase-9/ caspase-8 activation. Int J Mol Sci 2012; 13: 15523–35.
Boland K, Flanagan L, Prehn JH . Paracrine control of tissue regeneration and cell proliferation by Caspase-3. Cell Death Dis 2013; 11: 1–6.
Ma F, Zhang C, Prasad KV, Freeman GJ, Schlossman SF . Molecular cloning of Porimin, a novel cell surface receptor mediating oncotic cell death. Proc Natl Acad Sci U S A 2001; 98: 9778–83.
Varadarajan SG, An J, Novalija E, Stowe DF . Sevoflurane before or after ischemia improves contractile and metabolic function while reducing myoplasmic Ca2+ loading in intact hearts. Anesthesiology 2002; 96: 125–33.
Shuang Z, Hong LI, Shi JY . Tribulosin protects rat hearts from ischemia/reperfusion injury. Acta Pharmacol Sin 2010; 31: 671–8.
Zhu J, Rebecchi MJ, Tan M, Glass PS, Brink PR, Liu L . Age-associated differences in activation of Akt/GSK-3beta signaling pathways and inhibition of mitochondrial permeability transition pore opening in the rat heart. J Gerontol A Biol Sci Med Sci 2010; 65: 611–9.
Sperlagh B, Kittel A, Lajtha A, Vizi ES . ATP acts as fast neurotransmitter in rat habenula: neurochemical and enzyme cytochemical evidence. Neuroscience 1995; 66: 915–20.
Kordas KS, Sperlagh B, Tihanyi T, Topa L, Steward MC, Varga G, et al. ATP and ATPase secretion by exocrine pancreas in rat, guinea pig, and human. Pancreas 2004; 29: 53–60.
Zhang YW, Long C, Ding LL . Determination of phosphocreatine in muscular tissues by high performance liquid chromatography. Chin J Chromatogr 2001; 19: 251–2.
Liu L, Zhu J, Brink PR, Glass PS, Rebecchi MJ . Age-associated differences in the inhibition of mitochondria permeability transition pore opening by cyclosporine A. Acta Anaesthesiol Scand 2011; 55: 622–30.
Yao YT, Li LH, Chen L, Wang WP, Li LB, Gao CQ . Sevoflurane postconditioning protects isolated rat hearts against ischemia-reperfusion injury: the role of radical oxygen species, extracellular signal-related kinases 1/2 and mitochondrial permeability transition pore. Mol Biol Rep 2010; 37: 2439–46.
Tong JS, Zhang QH, Huang X, Fu XQ, Qi ST, Wang YP, et al. Icartin causes sustained ERK1/2 activation and induces apoptosis in human endometrial cancer cells. PLoS One 2011; 6: e 16781.
Harada H, Andersen JS, Mann M, Terada N, Korsmeyer SJ . p70S6 kinase signals cell survival as well as growth, inactivating the pro-apoptotic molecule BAD. Proc Natl Acad Sci U S A 2001; 98: 9666–70.
Di Lisa F, Ziegler M . Pathophysiological relevance of mitochondria in NAD+ metabolism. FEBS Lett 2001; 492: 4–8.
Enari M, Sakahira H, Yokoyama H, Okawa K, Iwamatsu A, Nagata S . A caspase-activated DNAse that degardes DNA during apoptosis, and its inhibitor ICAD. Nature 1998; 391: 43–50.
Majno G, Joris I . Apoptosis, oncosis and necrosis. An overview of cell death. Am J Pathol 1995; 146: 3–15.
Trump BF, Berezesky IK, Chang SH, Phelps PC . The pathways of cell death: oncosis, apoptosis and necrosis. Toxicol Pathol 1997; 25: 82–8.
Buja LM, Vela D . Cardiomyocyte death and renewal in the normal and diseased heart. Cardiovasc Pathol 2008; 17: 349–74.
Darzynkiewic Z, Juan G, Li X, Gorczyca W, Murakami T, Traganos F . Cytometry in cell necrobiology: analysis of apoptosis and accidental cell death (Necrosis). Cytometry 1997; 27: 1–20.
Acknowledgements
This work is supported by the National Natural Science Foundation of China (Grant No 81372024 to Dr Jiang ZHU), the Technology Bureau of Suzhou, China (Grants No SYS201130, SYSD2012085, and KJXW2011015 to Dr Chen WANG; SYS201125 and SYS201340 to Dr Jiang ZHU). Dr Chen WANG also received support from the Revitalizing the Key Talent's Subsidy Project in Science and Education (Jiangsu Province, China).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Xie, H., Zhang, J., Zhu, J. et al. Sevoflurane post-conditioning protects isolated rat hearts against ischemia-reperfusion injury via activation of the ERK1/2 pathway. Acta Pharmacol Sin 35, 1504–1513 (2014). https://doi.org/10.1038/aps.2014.78
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2014.78
Keywords
This article is cited by
-
Sevoflurane postconditioning protects against myocardial ischemia/reperfusion injury by restoring autophagic flux via an NO-dependent mechanism
Acta Pharmacologica Sinica (2019)
-
Sevoflurane Post-conditioning Protects Primary Rat Cortical Neurons Against Oxygen–Glucose Deprivation/Resuscitation: Roles of Extracellular Signal-Regulated Kinase 1/2 and Bid, Bim, Puma
Neurochemical Research (2015)
-
Investigation of therapeutic potential and molecular mechanism of vitamin P and digoxin in I/R-induced myocardial infarction in rat
Naunyn-Schmiedeberg's Archives of Pharmacology (2015)
