
Abstract
Aim:
Lapatinib is a dual inhibitor of EGFR and human epidermal growth factor receptor 2 (HER2), and used to treat advanced breast cancer. To overcome its poor water solubility, we constructed lapatinib-incorporated lipoprotein-like nanoparticles (LTNPs), and evaluated the particle characteristics and possible anti-breast cancer mechanisms.
Methods:
LTNPs (lapatinib bound to albumin as a core, and egg yolk lecithin forming a lipid corona) were prepared. The particle characteristics were investigated using transmission electron microscopy (TEM) and atomic force microscopy (AFM). The uptake and subcellular localization of LTNPs, as well as the effects of LTNPs on cell cycle were examined in BT-474 human breast cancer cells in vitro. Mice bearing BT-474 subcutaneous xenograft were intravenously injected with coumarin-6 loaded LTNPs (30 mg/kg) to study the targeting mechanisms in vivo.
Results:
The LTNPs particles were generally spherical but flexible under TEM and AFM, and approximately 62.1 nm in size with a zeta potential of 22.80 mV. In BT-474 cells, uptake of LTNPs was mediated by endosomes through energy-dependent endocytosis involving clathrin-dependent pinocytosis and macropinocytosis, and they could effectively escape from endosomes to the cytoplasm. Treatment of BT-474 cells with LTNPs (20 μg/mL) induced a significant cell arrest at G0/G1 phase compared with the same concentration of lapatinib suspension. In mice bearing BT-474 xenograft, intravenously injected LTNPs was found to target and accumulate in tumors, and colocalized with HER2 and SPRAC (secreted protein, acidic and rich in cysteine).
Conclusion:
LTNPs can be taken up into breast cancer cells through specific pathways in vitro, and targeted to breast cancer xenograft in vivo via enhanced permeability and retention effect and SPARC.
Similar content being viewed by others

Introduction
Breast cancer is the second leading cause of cancer death for women, and 20%–30% of these breast cancers overexpress human epidermal growth factor receptor 2 (HER2)1,2. Compared with HER2-negative breast cancer, HER2-positive cancers tend to be poorly differentiated and high grade, which often lead to lower patient survival rates3. Because HER2 plays a crucial role in the survival of cancer cells, it has become a therapeutic target for several drugs including monoclonal antibodies and small molecule kinase inhibitors.
Lapatinib (Tykerb, GlaxoSmithKline) is a dual tyrosine inhibitor of EGFR and HER2 that was approved by the US FDA for the treatment of HER2-positive breast cancer and has also showed positive results on many other cancers4,5,6. However, the poor water solubility of lapatinib restricted its clinical outcome. To improve the water solubility, we developed a type of surfactants-free nanoparticulated lapatinib by incorporating lapatinib into lipoprotein-like nanoparticles (LTNPs) based on the high binding efficiency (>99%) of lapatinib to blood albumin2,7. LTNPs could tremendously increase the water solubility of Lapatinib from 7 μg/mL to over 10 mg/mL. LTNPs have comparative anti-breast cancer effects at a cumulative dose of only 15% of the commercially available lapatinib tablet named Tykerb7. However, the targeting mechanism of LTNPs remained unclear.
Herein, we report several experiments aimed at elucidating the potential breast cancer targeting mechanism of LTNPs. To track the behavior of LTNPs, the fluorescent dye coumarin-6 was loaded into LTNPs. Subcellular location and uptake mechanisms were evaluated to determine how BT-474 cells take up LTNPs. Tumor sections were prepared and specific receptors were stained to demonstrate the in vivo tumor targeting mechanism of LTNPs.
Materials and methods
Materials
Lapatinib was purchased from Rongda Pharm & Chem Co, Ltd (Hangzhou, China). Bovine serum albumin (BSA) and coumarin-6 were purchased from Sigma (Saint Louis, MO, USA). Egg yolk lecithin (EYL) was purchased from Shanghai Advanced Vehicle Technology Pharmaceutical Ltd (Shanghai, China). Monensin, chlorpromazine, filipin, nacodazole, sodium azide and brefeldin A (BFA) were purchased from J&K Scientific Ltd (Beijing, China). Rabbit anti-HER2 IgG was purchased from Boster (Wuhan, China). The anti-secreted protein, acidic and rich in cysteine (SPARC) was purchased from Santa Cruz Biotechnology, Inc (Santa Cruz, CA, USA). DAPI was purchased from Beyotime (Haimen, China). Normal donkey serum, AMCA-conjugated affinipure donkey anti-sheep IgG and Cy3 conjugated affinipure donkey anti-rabbit IgG were purchased from Jackson ImmunoResearch Laboratories, Inc (West Grove, PA, USA). The human breast cancer cell line BT-474 was purchased from the Institute of Biochemistry and Cell Biology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences (Shanghai, China). RPMI1640 cell culture medium and fetal bovine serum were purchased from Gibco (Grand Island, NY, USA). Plastic cell culture dishes and plates were purchased from Wuxi NEST Biotechnology Co Ltd (Wuxi, China). All other chemicals were purchased from Sinopharm Chemical Reagent (Shanghai, China).
Animals
Female CB-17 SCID mice aged 4–6 weeks were obtained from Vitalriver (Beijing, China) and maintained at 22±2 °C on a 12 h light-dark cycle with free access to food and water. The animals used for the experiment were treated according to the protocols evaluated and approved by the Ethical Committee of Sichuan University.
Preparation of lapatinib nanoparticles
LTNPs were prepared according to the procedure described previously7. Briefly, 6 mg lapatinib was added into a mixture of 6 mL ethanol and 2 mL deionized water and then mixed with 2 mL dichloromethane containing 450 mg EYL. The mixture was dropped into 140 mL of agitated deionized water containing 150 mg BSA and stirred for 40 min. LTNPs could be observed after the organic solvents were removed with a rotary evaporator.
The coumarin-6-loaded LTNPs were prepared as above except with the addition of courmain-6 dissolved in the EYL solution at a concentration of 600 μg/mL.
Characterization of LTNPs
The mean particle size and zeta potential of LTNPs were determined by dynamic light scattering (DLS) using a zeta potential/particle sizer (380ZLS, PSS Nicomp Particle Size System, Sant Barbara, CA, USA). Samples were stained with 2% phosphotungstic acid and the morphology was observed using a transmission electron microscope (TEM; H-600, Hitachi, Tokyo, Japan). LTNPs were also charaterized by atomic force microscope (AFM; Multimode Scanning Probe Microscope, Digital Instrument, Santa Barbara, CA, USA)
Intracellular tracking of LTNP
BT-474 cells were seeded in glass-bottom dishes at the density of 1×104 per dish. After 24 h and 5 min of preincubation with PBS, cells were treated for 30 min with 100 μg/mL (referred to the concentration of lapatinib herein) coumarin-6 loaded LTNPs in Hank's balanced salt solution. Transferrin-Texas Red (100 μg/mL) was used to mark early and recycling endosomes, while LysoTracker Blue DND-22 (50 nmol/L) marked endolysosomal compartments. Cells were washed three times and then fixed and mounted in fluorescent mounting medium. To evaluate the release of LTNPs from endosomes, cells were pretreated with 100 μg/mL coumarin-6 loaded LTNPs for 30 min and then treated as above for another 30 min. Images were observed using confocal microscopy (TCS SP5, Leica, Wetzlar, Germany).
Uptake mechanism of LTNPs by BT-474 cells
BT-474 cells (2×105·mL−1·well−1) were seeded in 6-well plates. After 48 h, cells were pre-incubated for 20 min in Dulbecco's modified Eagle medium and treated for 30 min with 500 μg/mL of LTNPs and various inhibitors, which were PBS (control), 10 μg/mL filipin, 450 mmol/L sucrose, 0.1% w/v sodium azide, 10 mmol/L ammonium chloride, 100 nmol/L monensin, 33 μmol/L nacodazole, 25 μmol/L cytochalasin D, 20 μg/mL BFA and 20 μg/mL chlorpromazine. After five washes with ice-cold PBS, the cells were harvested and resuspended in 0.5 mL PBS. The mean fluorescence intensity was observed by flow cytometry (FACS Aria Cell Sorter, BD, San Diego, CA, USA).
Cell cycle assay
The BT-474 cells were seeded in 6-mm dishes at a density of 1×105 cells/mL. Twenty four hours later, the culture media in the dishes was replaced with fresh culture media containing either 20 μg/mL lapatinib suspension (LTS) or 20 μg/mL LTNPs. After an additional 24 h, cells were harvested and fixed in 70% ethanol for 24 h. The fixed cells were resuspended in 0.5 mL PBS containing propiumiodide (50 μg/mL) and RNase (100 μg/mL) and incubated at 37 °C for 30 min. A cell cycle assay was performed using FACS (BD, San Diego, CA, USA).
Tumor distribution
Mice bearing BT-474 xenografts were established as described previously8. Briefly, 2×106 BT-474 cells/100 μL was injected into the right flank of a female CB-17 SCID mouse. Tumor length and width were measured every 2 d with a caliper and the tumor volume was calculated from the following equation: (length×width2)/2. When the tumor reached 50 to 100 mm3, the mouse was intravenously injected with 30 mg/kg coumarin-6 loaded LTNPs. Two hours later, the mouse was anesthetized and the heart was perfused with saline followed by 4% paraformaldehyde. The tumor was then removed and prepared in frozen sections using a cryotome Cryostat (CM 1900, Leica, Solms, Germany). The nuclei were stained with 1 μg/mL DAPI. Fluorescent distribution was captured with a confocal microscope (TCS SP5, Leica, Solms, Germany).
Fluorescent in situ hybridization of BT-474 tumor slides
Mice bearing BT-474 xenografts were injected with coumarin-6 loaded LTNPs. Two hours later, frozen and sectioned tumors were prepared as above and stained as described previously9. After incubation with 0.5% Triton X-100 in pH 7.2 Tris buffer for 2 h at room temperature, sections were blocked with 5% donkey serum for 2 h. Sections were further incubated with rabbit anti-human HER2 and sheep anti-human SPARC overnight at 4 °C. Sections were rinsed three times with TBS and incubated for 2 h at room temperature with AMCA conjugated donkey anti-rabbit secondary antibody and Cy3 conjugated donkey anti-sheep secondary antibody. Sections were rinsed three times with TBS and were covered with a glass coverslip. The fluorescent distribution was captured with a confocal microscope (TCS SP5, Leica, Germany).
Results
Characterization
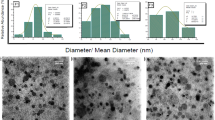
The mean particle size determined by DLS was 62.1 nm (Figure 1A) with a polydispersity index of 0.21. The Zeta potential was 22.80±8.13 mV. Through transmission electronic microscopy and atomic force microscopy, the particles were identified as general vesicles (Figure 1B and 1C). Though three-dimensional reconstruction using AFM showed that the LTNPs had a spherical shape, particle heights did not correlate with the relative diameters. The diameters were approximately 60 nm and the heights were less than 3 nm (Figure 1D and 1E), demonstrating that EYL was flexible, which is the major component of LTNP.

LTNPs particle size was characterized by DLS (A), TEM (B) and AFM (C, D, and E). The bar of TEM was 100 nm. The results of AFM are shown by 3D reconstruction (C), phase diagram (D) and height curve (E).
Intracellular tracking
After 30 min incubation, LTNPs not only colocalized with early endosomes and recycling endosomes but also apparently colocalized with secondary endosomes and endosomes (Figure 2D). LTNPs were also distributed throughout the cytoplasm, indicating that LTNPs were uptaken by early endosomes and then into secondary endosomes occurred quickly and that LTNPs could be released into the cytoplasm. After 1-h incubation, there was no colocalization of LTNP and second endosomes and endosomes, indicating that LTNPs were mostly released into the cytoplasm (Figure 2H). These results suggested that LTNPs could escape from the endosomes into the cytoplasm, which was consistent with studies in other cells10.

Intracellular tracking of LTNPs. BT-474 cells incubated with LTNPs for 30 min (A, B, C and D) or 1 h (E, F, G and H). Late endosomes stain with LysoTracker Blue (A and E). Coumarin-6-loaded LTNPs are green (B and F). Early endosomes stain by Transferrin-Texas Red (C and G). The merged pictures are also shown (D and H).
Uptake mechanism
The uptake of LTNPs by BT-474 cells occurred through energy-dependent endocytosis because of uptake was reduced to 88.4% of the control after energy depletion by sodium azide (Figure 3). Several mechanisms are involved in endocytosis: caveolae-mediated endocytosis, clathrin-mediated endocytosis, macropinocytosis and clathrin and caveolae-independent endocytosis11,12. Sucrose and chlorpromazine (an agent that blocks the formation of clathrin-coated pits) decreased the uptake to 88.5% and 79.0%, respectively, suggesting the involvement of clathrin-mediated endocytosis, which was in agreement with a previous study10. Filipin, a special inhibitor of caveolae-associated endocytosis13, elevated the uptake to 136%, indicating that the uptake of LTNPs was not mediated by caveolae-associated endocytosis. Nocodazole significantly decreased the uptake to 65.2%, suggesting that macropinocytosis was also involved in the uptake.

Uptake mechanism of coumarin-6 loaded LTNPs by BT-474 cells. bp<0.05 compared with control.
On the other hand, the uptake may have involved acid endosomes because monensin, an antiporter of Na+/H+, reduced the uptake to 56.8%. The Golgi apparatus also participated in the uptake procedure, as the uptake was reduced to 74.6% after the Golgi apparatus was destroyed by the addition of BFA.
Cell cycle assay
The BT-474 control cells were mostly distributed in the G0/G1 phase. Only 24.71% of cells were in the S phase, while 12.68% were in the G2/M phase. After treatment with LTS and LTNPs, the proportion of cells in G0/G1 phase was significantly increased and the proportion of cells in G2/M phases was significantly decreased (Figure 4 and Table 1). Meanwhile, the number of cells that were in G0/G1 phase after treatment with LTNPs was significantly higher than that of cells treated with LTS. The number of cells in G2/M phase after treatment with LTNPs was significantly lower than that of cells treated with LTS.

Cell cycle assay of BT-474 cells treated with control (A), 20 μg/mL of LTS (B) or 20 μg/mL of LTNPs (C) for 24 h.
Tumor distribution
The crowded nuclei demonstrated the quick growth of the BT-474 tumor. LTNPs were distributed throughout the whole site around the nuclei (Figure 5), demonstrating that LTNPs could effectively target and accumulate in tumors. This was consistent with a study performed on U87 glioma tumor-bearing mice5.

Distribution of LTNPs in a subcutaneous BT-474 tumor slide. Nuclei are stained blue by DAPI (A), LTNPs are displayed in green (B) and the merged picture is shown (C).
Fluorescent in situ hybridization
Fluorescence in situ hybridization demonstrated a high expression level of both HER2 and SPARC (Figure 6). This observation was consistent with previously published results14. The LTNPs colocalized with SPARC, indicating that SPARC may be a target of LTNPs.

Expression of HER2 and SPARC in BT-474 tumor slices treated with coumarin-6-loaded LTNPs. Blue is HER2, green is LTNP and red is SPARC. The merged picture is also shown.
Discussion
Nanotechnology is a promising method in drug development. Since the first nanoformulation-liposomes were developed in the mid-twentieth century, only a few nanomedicines such as Doxil have become commercially available15. The complex preparation procedures and toxicity of materials are two huge challenges in the progression of nanomedicine from laboratory research to clinical applications16,17. In this study, a convenient procedure with injectable materials was developed. As demonstrated previously, the LNTPs structure was similar to that of lipoprotein, including a lipid surface layer and a core of conjugation of BSA and lapatinib7. As most or all of the surface of LTNPs was composed of EYL, the particles were stickier than other nanoparticles like liposomes, which were identified by AFM. The AFM-calculated particle size was approximately 60 nm, while the height of the particles was less than 5 nm.
The uptake of LTNPs by BT-474 cells was time- and concentration-dependent, though the exact uptake mechanism remained unclear. It was shown that the uptake of compounds into cells could be mediated by active endocytosis and passive diffusion. Due to the large scale of nanoparticles, the involvement of passive diffusion was unlikely. In this study, the uptake largely decreased after energy depletion by sodium azide, demonstrating that the uptake was due to energy-dependent endocytosis. Although there are at least four mechanisms involved in endocytosis, our study suggested only clathrin-mediated endocytosis and macropinocytosis participated in the uptake procedure. Macropinocytosis evidently occurred between cells and particles larger than 100 nm. Although the average LNTPs particle size was only 62.1 nm, some particles were measured over 100 nm18, which might be the reason that macropinocytosis was involved in the uptake. Clathrin-dependent pinocytosis is used by all eukaryotic cells to internalize nutrients and degrade or recycle substances. Thus, this process is well-known for its role in the selective uptake of molecules mediated by specific receptors18. Several studies suggested that BSA nanoparticles could be recognized by SPARC. Since BSA is a main component of our particles, SPARC may be a target of LTNPs. Through fluorescent in situ hybridization, SPARC expression in the tumor slice was found to colocalize with LTNPs, suggesting the involvement of SPARC in the uptake of LTNPs.
To inhibit the survival (phosphatidylinositol 3-kinase/Akt) pathways and induce apoptosis, lapatinib could bind to the tyrosine domain of EGFR and HER22. The phosphatidylinositol 3-kinase/Akt pathway is essential for cell proliferation, and blocking this pathway could lead to cellular arrest in the G1/S phase. In our study, LTS arrested more cells in the S phase and G0/G1 phase, which is consistent with previous findings10. LTNPs arrested even more cells in the G0/G1 phase compared to LTS, suggesting that LTNPs were more effective at inhibiting tumor cell proliferation. This was consistent with our previous studies7,10.
Tumor slices were prepared to further elucidate the in vivo effect of LTNPs. The results demonstrated that LTNPs were effectively distributed throughout the tumor. The quick amplification of tumor cells leads to a leaky vasculature in the tumor, which makes it more permeable; this is better known as the enhanced permeability and retention (EPR) effect19,20,21,22. The cut-off size of the permeabilized vasculature varies from case to case and the size of carriers may largely affect the effectiveness of drug delivery23. Normally, the cut-off size of subcutaneous tumor is higher than 500 nm. With a particle size of only 62.8 nm, LTNPs could utilize the EPR effect to target tumor cells and accumulate in BT-474 xenografts. On the other hand, SPARC is a candidate of LTNPs receptor, as discussed above. The expression of SPARC in tumors may also contribute to the accumulation of LTNPs in tumors.
Conclusion
A 62.8-nm LTNPs that incorporated lapatinib was prepared. LTNPs were taken up into BT-474 cells through endosomes and could effectively escape from the endosomes to the cytoplasm. Furthermore, the uptake was mediated by energy-dependent endocytosis, which involved clathrin-dependent pinocytosis and macropinocytosis. LTNPs could induce the arrest of BT-474 cells in G0/G1 phase at significantly higher levels compared to LTS. In vivo, LTNPs could also target subcutaneous BT-474 xenografts and accumulate in the tumors through the EPR effect and SPARC mediated targeting.
Author contribution
Hui-le GAO and Qin HE designed the study; Li ZHANG performed the experiments and wrote the paper with the assistance of Shuang ZHANG, Shao-bo RUAN and Qian-yu ZHANG.
References
Moreira C, Kaklamani V . Lapatinib and breast cancer: current indications and outlook for the future. Expert Rev Anticancer Ther 2010; 10: 1171–82.
Medina PJ, Goodin S . Lapatinib: a dual inhibitor of human epidermal growth factor receptor tyrosine kinases. Clin Ther 2008; 30: 1426–47.
Burstein HJ . The distinctive nature of HER2-positive breast cancers. N Engl J Med 2005; 353: 1652–54.
LaBonte MJ, Wilson PM, Fazzone W, Russell J, Louie SG, El-Khoueiry A, et al. The dual EGFR/HER2 inhibitor lapatinib synergistically enhances the antitumor activity of the histone deacetylase inhibitor panobinostat in colorectal cancer models. Cancer Res 2011; 71: 3635–48.
Gao H, Yang Z, Cao S, Xi Z, Zhang S, Pang Z, et al. Behavior and anti-glioma effect of lapatinib-incorporated lipoprotein-like nanoparticles. Nanotechnology 2012; 23: 435101.
Olaussen KA, Commo F, Tailler M, Lacroix L, Vitale I, Raza SQ, et al. Synergistic proapoptotic effects of the two tyrosine kinase inhibitors pazopanib and lapatinib on multiple carcinoma cell lines. Oncogene 2009; 28: 4249–60.
Gao H, Cao S, Chen C, Cao S, Yang Z, Pang Z, et al. Incorporation of lapatinib into lipoprotein-like nanoparticles with enhanced water solubility and anti-tumor effect in breast cancer. Nanomedicine (Lond) 2013; 8: 1429–42.
Heikkila JE, Vaha-Koskela MJ, Ruotsalainen JJ, Martikainen MW, Stanford MM, McCart JA, et al. Intravenously administered alphavirus vector VA7 eradicates orthotopic human glioma xenografts in nude mice. PLoS One 2010; 5: e8603.
Gao H, Pan S, Yang Z, Cao S, Chen C, Jiang X, et al. A cascade targeting strategy for brain neuroglial cells employing nanoparticles modified with angiopep-2 peptide and EGFP-EGF1 protein. Biomaterials 2011; 32: 8669–75.
Gao H, Wang Y, Chen C, Chen J, We Y, Cao S, et al. Incorporation of lapatinib into core-shell nanoparticles improves both the solubility and anti-glioma effects of the drug. Int J Pharm 2014; 461: 478–88.
Liu J, Shapiro JI . Endocytosis and signal transduction: basic science update. Biol Res Nurs 2003; 5: 117–28.
Gao H, Yang Z, Zhang S, Cao S, Shen S, Pang Z, et al. Ligand modified nanoparticles increases cell uptake, alters endocytosis and elevates glioma distribution and internalization. Sci Rep 2013; 3: 2534.
Lu W, Sun Q, Wan J, She Z, Jiang XG . Cationic albumin-conjugated pegylated nanoparticles allow gene delivery into brain tumors via intravenous administration. Cancer Res 2006; 66: 11878–87.
Zhang LM, Zhang JN, Yang SW, Dong C, Fang DD, Li J, et al. Expression of HER2 in glioma cell lines A172, U251, U87 and SHG-44 and its significance. Chin J Neurosurg Dis Res 2011; 10: 151–53.
Barenholz Y . Doxil®—The first FDA-approved nano-drug: Lessons learned. J Control Release 2012; 160: 117–34.
Gao H, Pang Z, Pan S, Cao S, Yang Z, Chen C, et al. Anti-glioma effect and safety of docetaxel-loaded nanoemulsion. Arch Pharm Res 2012; 35: 333–41.
Gao H, Pang Z, Jiang X . Targeted delivery of nano-therapeutics for major disorders of the central nervous system. Pharm Res 2013; 30: 2485–98.
Mahmoudi M, Azadmanesh K, Shokrgozar MA, Journeay WS, Laurent S . Effect of nanoparticles on the cell life cycle. Chem Rev 2011; 111: 3407–32.
Maeda H . SMANCS and polymer-conjugated macromolecular drugs: advantages in cancer chemotherapy. Adv Drug Deliv Rev 2001; 46: 169–85.
Matsumura Y, Maeda H . A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res 1986; 46: 6387–92.
Torchilin VP . Passive and active drug targeting: drug delivery to tumors as an example. Handb Exp Pharmacol 2010; (197): 3–53.
Fang J, Sawa T, Maeda H . Factors and mechanism of “EPR” effect and the enhanced antitumor effects of macromolecular drugs including SMANCS. Adv Exp Med Biol 2003; 519: 29–49.
Hobbs SK, Monsky WL, Yuan F, Roberts WG, Griffith L, Torchilin VP, et al. Regulation of transport pathways in tumor vessels: role of tumor type and microenvironment. Proc Natl Acad Sci U S A 1998; 95: 4607–12.
Acknowledgements
The work was funded by the National Natural Science Foundation of China (81373337) and the Sichuan University Starting Foundation for Young Teachers (2014SCU11044).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Zhang, L., Zhang, S., Ruan, Sb. et al. Lapatinib-incorporated lipoprotein-like nanoparticles: preparation and a proposed breast cancer-targeting mechanism. Acta Pharmacol Sin 35, 846–852 (2014). https://doi.org/10.1038/aps.2014.26
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2014.26
Keywords
This article is cited by
-
Application of atomic force microscopy in cancer research
Journal of Nanobiotechnology (2018)
-
Co-delivery of docetaxel and silibinin using pH-sensitive micelles improves therapy of metastatic breast cancer
Acta Pharmacologica Sinica (2017)
-
Silibinin and indocyanine green-loaded nanoparticles inhibit the growth and metastasis of mammalian breast cancer cells in vitro
Acta Pharmacologica Sinica (2016)
