
Abstract
Bile acids (BAs) are traditionally considered as “physiological detergents” for emulsifying hydrophobic lipids and vitamins due to their amphipathic nature. But accumulating clinical and experimental evidence shows an association between disrupted BA homeostasis and various liver disease conditions including hepatitis infection, diabetes and cancer. Consequently, BA homeostasis regulation has become a field of heavy interest and investigation. After identification of the Farnesoid X Receptor (FXR) as an endogenous receptor for BAs, several nuclear receptors (SHP, HNF4α, and LRH-1) were also found to be important in regulation of BA homeostasis. Some post-translational modifications of these nuclear receptors have been demonstrated, but their physiological significance is still elusive. Gut secrets FGF15/19 that can activate hepatic FGFR4 and its downstream signaling cascade, leading to repressed hepatic BA biosynthesis. However, the link between the activated kinases and these nuclear receptors is not fully elucidated. Here, we review the recent literature on signal crosstalk in BA homeostasis.
Similar content being viewed by others


Introduction
Cholesterol is directly converted by a series of chemical reactions into more soluble amphipathic primary bile acids (BAs) in hepatocytes. Therefore, the BA biosynthesis pathway is the primary route of cholesterol disposal from the body1. Through specialized hepatic transporters, hepatic BAs are then transported into bile canaliculi, which carry them into the gallbladder, where bile is concentrated. After each meal, bile is emptied from the gallbladder and, due to its amphipathic nature, emulsifies lipids and hydrophobic vitamins to facilitate their intestinal absorption. Excess BAs are reabsorbed by the ileum, which is the distal part of the intestine, and travel back to hepatocytes through the portal vein. Up to 95% of BAs will be reabsorbed in this process, which is fittingly termed the “enterohepatic recycle of BAs.”
A number of diseases cause cholestasis, a condition in which bile fails to flow out of the liver. Cholestasis usually induces chronic or acute liver toxicity and damage2. Furthermore, BA dysregulation has been associated with a variety of liver and metabolic diseases, other than those directly affected by blocked BA transport. Hepatitis B viruses (HBVs) enter hepatocytes through the transporter for BA uptake, namely the Na+-taurocholate cotransporting polypeptide (NTCP)3. HBV infection causes increased BA biosynthesis4. When added to the existing type 2 diabetes treatment regimens, BA sequestrants, which bind BAs to prevent their ileal reabsorption, exhibited beneficial effects for these patients5. BA alteration is also associated with obesity, and increased circulating BAs were observed in patients who had undergone bariatric surgeries6,7. Elucidating the regulatory mechanisms behind BA homeostasis could allow for the identification of new therapeutic targets useful for improved treatment of relevant diseases.
In BA biosynthesis, p450 family member cholesterol 7α-hydroxylase (Cyp7a1) is the rate-limiting enzyme for the conversion of cholesterol to BAs8,9. Further demonstration of the functional significance of Cyp7a1 comes from the loss-of-function mutation in humans, which results in a hypercholesterolemic phenotype10. With the cloning of Cyp7a1, the feedback regulation of Cyp7a1 transcription was also observed as excess BAs inhibited Cyp7a1 transcription11. Given the functional importance of Cyp7a1 in both cholesterol and BA homeostasis, the transcriptional regulation of Cyp7a1 has been the attention of research in the field for over 20 years1 and a number of regulatory factors have been identified. Specifically, identification of the Farnesoid X Receptor (FXR, also known as NR1H4) as the endogenous receptor for BAs12,13 has ushered our understanding of BA biosynthesis, at both molecular and physiological levels, into a new era.
Before the identification of FXR, BAs were never considered anything more than “physiological detergents.” However, accumulating data has led to the designation of BAs as important “signaling hormones”14. Inside the intestine, BAs are further modified into structurally diverse secondary BA species by gut microbiota15. More intriguingly, different primary and secondary BA species exhibit different capacities to bind and activate FXR, making BA composition a crucial factor for determining FXR activity in various tissues15,16. Within hepatocytes, BAs bind FXR, and together they transactivate the small heterodimer partner (SHP, also known as NR0B2). SHP represses Cyp7a1 transcription by binding with two other nuclear receptors, including liver receptor homolog-1 (LRH-1, also known as NR5A2) and hepatocyte nuclear factor 4α (HNF4α, also known as NR2A1), both of which are required for the basal transactivation of Cyp7a117,18,19,20. The inhibition of Cyp7a1 by the BA-FXR-SHP axis resonates with the feedback regulation of Cyp7a1 transcription by excess BAs, which provided the molecular basis for the intrahepatic feedback regulation of BA biosynthesis. Despite the complex phenotypes, including hypertriglyceridemia, the analysis of FXR knockout (KO) mice further supported this model, as FXR KO mice displayed elevated BA levels, decreased SHP expression, and increased Cyp7a1 expression21. However, SHP KO mice only exhibited a mild increase in BA and were still responsive to feedback regulation of BA biosynthesis22. Moreover, BA synthesis and Cyp7a1 expression are dramatically higher in mice with double knockout (DKO) of FXR and SHP than with either gene alone23. Although these observations were still considered consistent with the intrahepatic feedback model, they also allow for additional pathway or factor involvement in the tight control of BA biosynthesis.
The ileum has long been recognized as the site for BA absorption24,25, which is physiologically indispensable for the intrahepatic feedback model. It was a surprising discovery that the ileum acts as more than simply a canal for BA recycling26. In ileocytes, BAs bind to FXR and transactivate fibroblast growth factor 19 (FGF19, FGF15 as the mouse ortholog)26. Then, FGF15/19, specially expressed in the ileum27, binds to FGF receptor 4 (FGFR4), which activates a cascade of mitogen-activated protein (MAPK) kinase signaling26,28,29. Accordingly, FGF15 or FGFR4 KO mice exhibited increased BA levels26,30. Regulators of this pathway were also proven to be essential players in BA biosynthesis. For example, FGF15-interacting protein Diet, which is also specifically expressed in intestine, regulates FGF15 expression. Therefore, Diet regulates hepatic Cyp7a1 expression and lipid homeostasis31. Shp2, which is a cytoplasmic tyrosine phosphatase with two SH2 domains, positively regulates FGFR4 and was proven to be indispensable for the regulation of hepatic BA biosynthesis and Cyp7a1 expression32. Undoubtedly, the FGF19/FGFR4 pathway constitutes another feedback loop for repression of BA biosynthesis.
The ileum is responsible for eliciting two feedback signals: BAs and FGF19/15. BAs, together with FXR, provide intrahepatic feedback regulation of Cyp7a1 expression. Meanwhile, FGF19/15, the ligand for FGFR4, is secreted from the ileum, further providing an extrahepatic feedback signal for Cyp7a1 repression. Overall, the research on these two feedback branches remains two parallel paths. However, several studies implied the intertwining of these two pathways. In FXR or SHP KO mice, at the very least, impaired Cyp7a1 expression followed FGF19/15 injection26,33. In turn, in FGFR4 or FGF15 KO mice, FXR agonists failed to repress Cyp7a1 expression26,33. Also, in the mice with Shp2 deleted in hepatocytes, several lines of evidence suggested the cross-regulation of these two pathways32. Despite increased BAs and FGF15, the activation of both pathways was impaired in this mouse line. Furthermore, failed activation of FXR by a synthetic FXR agonist was also detected in the same animals with uncompromised expression and subcellular localization of FXR32. All of these studies, albeit not directly, point to the possible regulation of bile acid synthesis beyond transcription and translation levels.
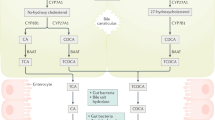
The post-translational regulation of nuclear receptors (FXR, SHP, HNF4α, and LRH-1) has been highlighted in the literature, but the physiological significance of this regulation remains inconclusive34. Due to metabolic roles of these nuclear receptors in BA homeostasis, these receptors are well suited for therapeutic targeting16,35. In this review, we summarize the functional phosphorylation sites of these nuclear receptors, sites that alter the transactivity of these receptors in vitro, and discuss the possible physiological implications of these receptors in BA biosynthesis (Figure 1).

Summary of possible kinases in the regulation of nuclear receptors in bile acid homeostasis. The confirmed substrates for specific kinases (at least by in vitro assays) are indicated with solid arrows, whereas proteins merely implicated to be the substrates of specific kinases are shown with dashed arrows.
FXR
In human hepatocytes, activating FXR via BAs induces FGF19 expression36,37. Therefore, FGFR4 downstream kinase Erk1/2 is activated by BA treatments37. Inhibition of FGF19 by neutralizing antibodies or FGFR4 activity by siRNA knockdown abrogates inhibition of Cyp7A1 by FXR agonist GW4064 in human hepatocytes, further supporting FGFR4-dependent FXR activity37.
FXR was first shown to be phosphorylated at serine 135 and 154 by protein kinase C (PKC) α and βI38. Mutation of both serines led to reduced FXR transcription activity without impairment of its DNA-binding capacity38. BAs are able to activate several isoforms of PKC in cultured cell lines and in vitro kinase assays39,40,41. In another study, a distinct PKC isoform, PKCζ, could phosphorylate FXR at threonine 456 to prevent cholestasis42. Despite the completely different PKC isoforms and phosphorylation sites, both studies indicated a possible role of PKC activation in the feedback regulation of BA biosynthesis mediated by BAs. More recently, PKCζ was shown to be activated by FGF19 in both cultured hepatocytes and in mouse livers43. Although FGF19 failed to induce PKCζ in another study, specific phosphorylation of other PKC isoforms was detected32.
In vitro data showed that activated PKC could phosphorylate FXR and enhance its transcription activity38. However, careful studies are still needed to establish PKC's role in BA homeostasis. First, FXR mutations at phosphorylation sites have to exhibit defective activity in vivo either in individuals carrying such mutations or knockin models. Second, the isoform(s) of PKC need(s) to be characterized in experimental models. Third, if PKC isoform(s) are involved in BA homeostasis, how PKC isoform(s) are activated under physiological or pathological conditions still requires further investigation.
Another possible kinase for regulation of BA biosynthesis is the AMP-activated protein kinase (AMPK). AMPK can be activated by BAs for establishing hepatocyte polarization44. Moreover, metformin, activator of AMPK, protects BA-induced hepatocyte apoptosis45. Although both studies investigated other important aspects of BA regulatory functions, they highlighted the possible role of AMPK in BA biosynthesis. A recent study reported that AMPK could phosphorylate FXR in vivo at serine 25046. More importantly, in both in vitro and in vivo models, AMPK activators can antagonize BA- or FXR agonist-induced SHP expression46. Given the important roles of AMPK in lipid and glucose metabolism, AMPK shows functional relevance with both BA/FXR and FGF19/FGFR4 signaling. Elucidation of AMPK's roles in FXR regulation could shed new light on the fine-tuning of this important signaling pathway.
SHP
The transcription of SHP is dynamic, possibly because BA biosynthesis is regulated by nutrient supply and the circadian clock47,48. Given that the transcription of SHP is regulated by the circadian clock47, the stability of its protein is low49. Erk can phosphorylate SHP in a BA- and FGF19-dependent manner49. This is the first time that FGF19/FGFR4 and BA/FXR signaling converge in the regulation of a specific Cyp7a1 regulator. FGF19/15 could mildly induce SHP expression26,32 in some studies and failed to induce any SHP expression in other reports. This could be due to subtle experimental design differences such as sample collection timing. However, they further argued for additional factor(s) in the regulation of BA biosynthesis, especially because SHP knockout mice showed only mild defects in BA biosynthesis regulation22.
More recently, the same group that reported SHP phosphorylation identified another phosphorylation site (threonine 55) on SHP43. This phosphorylation site was required for SHP binding with HNF4α and LRH-1, resulting in the failure to repress Cyp7a1 by BA and FGF19 signals. Of note, this site was phosphorylated by PKCζ, the activation of which by BA and FGF19 signals still needs further independent confirmation. Still, a better approach for evaluating these sites is to establish knockin mouse lines, especially when the SHP knockout mice exhibited only mild defects of BA biosynthesis regulations.
HNF4α
HNF4α is a definite regulator of Cyp7a1 and binds to the Cyp7a1 promoter for its basal activation. HNF4α is also required for the repression of Cyp7a1 through its association with SHP. However, its binding to the Cyp7a1 promoter does not change with FGFR activation by FGF19. Mass spectrometry studies have shown HNF4α can be phosphorylated at several serine/threonine sites50,51, suggesting that phosphorylation of HNF4α is an important route for regulating its activity.
Glucagon and cAMP were able to repress Cyp7a1 expression52. Increased HNF4α phosphorylation by protein kinase A (PKA) consequently reduced HNF4α binding to the Cyp7a1 promoter52. Although only PKA was the focus in this study, the possible roles of AMPK in the regulation of HNF4α during Cyp7a1 transcriptional regulation cannot be ruled out. Furthermore, not only can HNF4α be phosphorylated by AMPK but its dimerization and stability can also be reduced by the phosphorylation53. It should be noted that both FXR and HNF4α could be the targeted BA biosynthesis regulators by AMPK.
Due to the pleiotropic functions of HNF4α in various metabolic processes, multiple kinases have been shown to phosphorylate HNF4α under different conditions, which include mitogen-activated protein kinase p3854, Erk55, Src tyrosine kinase56, JNK57, and PKC58. Erk activation downstream of FGFR4 was proven to be indispensable for the repression of Cyp7a126,32,33,48. Among these kinases, Erk is the only one that is undoubtedly activated by FGF15/19. It would be interesting to examine the possible phosphorylation of HNF4α by both FGF19 and BA signals to determine whether the phosphorylation is involved in the direct switch of the downstream target in the feedback regulation of BA biosynthesis.
LRH-1
LRH-1 is another definite regulator of Cyp7a1 transcription, which binds with HNF4α and SHP at the Cyp7a1 promoter. Hepatocyte-specific knockout of LRH-1 showed that regulation of BA homeostasis by LRH-1 is not essential for feedback regulation of BA synthesis19. Erk can phosphorylate LRH-1 in vitro at serines 238 and 243 in the hinge domain, which increases the transcription activity of LRH-159.
Shp2 and regulation of BA-related kinases
As discussed above, FGF15/19/FGFR4 represents an extracellular mechanism for the repression of BA biosynthesis and BA/FXR signaling is the main player in the nuclei. The post-translational modification is an appealing mechanism to bridge these two spatially restricted pathways. Recent work on the cytoplasmic protein tyrosine phosphatase has shed new light on the intersection of these two pathways. In mice with Shp2 deleted in hepatocytes (Shp2hep−/−), BA biosynthesis is uncontrollably high with both FGF15/FGFR4 and FXR signaling exhibited defective activation, despite the high expression of the respective ligands32.
Detailed BA composition analysis revealed that contrary to the dramatic increase in the FXR agonist species, the FXR antagonist species remained unchanged in Shp2hep−/− mice. Without changes in expression, the impaired FXR activation could be due to post-translational changes in the protein or changes in co-activators. This observation was confirmed when synthetic FXR agonist treatment failed to activate FXR in Shp2hep−/− liver.
In the same mice, increased FGF15 expression also failed to activate FGFR4, as evidenced by lower Erk phosphorylation in the Shp2hep−/− liver. Shp2 binds with the direct target of FGFR4, FRS2α and is required for activation of the receptor. Defective activation of FGFR4 resulted in not only impaired Erk activation but also defective activation of multiple kinases, including p90RSK and PKC isoforms.
The results from Shp2hep−/− mice further proved that both the extrahepatic FGF15/19 signal and nuclear BA/FXR signal are orchestrated in hepatocytes by a cascade of signaling events. Furthermore, this model provided experimental evidence of crosstalk between these two pathways.
Perspectives
BAs have penetrated all areas of liver diseases as well as some gastrointestinal diseases. Given the toxicity of excess BAs, complicated regulatory circuits for BA homeostasis maintenance are necessary. Recently, two main repression signals for BA biosynthesis, namely BAs themselves and FGF15/19, have gained full recognition. However, the means by which these two signals are integrated inside hepatocytes still need further research. Post-translational modifications for nuclear receptors could provide the link for a fully integrated view of this important biological process. A more thorough understanding of the functional diversity of these nuclear receptors and their post-translational modifications is required to target them for future therapeutics.
References
Chiang JY . Bile acids: regulation of synthesis. J Lipid Res 2009; 50: 1955–66.
Gossard AA, Talwalkar JA . Cholestatic liver disease. Med Clin North Am 2014; 98: 73–85.
Yan H, Zhong G, Xu G, He W, Jing Z, Gao Z, et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife 2012; 1: e00049.
Oehler N, Volz T, Bhadra OD, Kah J, Allweiss L, Giersch K, et al. Binding of hepatitis B virus to its cellular receptor alters the expression profile of genes of bile acid metabolism. Hepatology 2014; 60: 1483–93.
Wang X, Fu X, Van Ness C, Meng Z, Ma X, Huang W . Bile acid receptors and liver cancer. Curr Pathobiol Rep 2013; 1: 29–35.
Myronovych A, Kirby M, Ryan KK, Zhang W, Jha P, Setchell KD, et al. Vertical sleeve gastrectomy reduces hepatic steatosis while increasing serum bile acids in a weight-loss-independent manner. Obesity 2014; 22: 390–400.
Ryan KK, Tremaroli V, Clemmensen C, Kovatcheva-Datchary P, Myronovych A, Karns R, et al. FXR is a molecular target for the effects of vertical sleeve gastrectomy. Nature 2014; 509: 183–8.
Chiang JY . Bile acid metabolism and signaling. Compr Physiol 2013; 3: 1191–212.
Lefebvre P, Cariou B, Lien F, Kuipers F, Staels B . Role of bile acids and bile acid receptors in metabolic regulation. Physiol Rev 2009; 89: 147–91.
Pullinger CR, Eng C, Salen G, Shefer S, Batta AK, Erickson SK, et al. Human cholesterol 7a-hydroxylase (CYP7A1) deficiency has a hypercholesterolemic phenotype. J Clin Invest 2002; 110: 109–17.
Jelinek DF, Andersson S, Slaughter CA, Russell DW . Cloning and regulation of cholesterol 7 alpha-hydroxylase, the rate-limiting enzyme in bile acid biosynthesis. J Biol Chem 1990; 265: 8190–7.
Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM, Luk A, et al. Identification of a nuclear receptor for bile acids. Science 1999; 284: 1362–5.
Parks DJ, Blanchard SG, Bledsoe RK, Chandra G, Consler TG, Kliewer SA, et al. Bile acids: natural ligands for an orphan nuclear receptor. Science 1999; 284: 1365–8.
Houten SM, Watanabe M, Auwerx J . Endocrine functions of bile acids. EMBO J 2006; 25: 1419–25.
Sayin SI, Wahlstrom A, Felin J, Jantti S, Marschall HU, Bamberg K, et al. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab 2013; 17: 225–35.
Thomas C, Pellicciari R, Pruzanski M, Auwerx J, Schoonjans K . Targeting bile-acid signalling for metabolic diseases. Nat Rev Drug Discov 2008; 7: 678–93.
Kir S, Zhang Y, Gerard RD, Kliewer SA, Mangelsdorf DJ . Nuclear receptors HNF4alpha and LRH-1 cooperate in regulating Cyp7a1 in vivo. J Biol Chem 2012; 287: 41334–41.
Lu TT, Makishima M, Repa JJ, Schoonjans K, Kerr TA, Auwerx J, et al. Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol Cell 2000; 6: 507–15.
Lee YK, Schmidt DR, Cummins CL, Choi M, Peng L, Zhang Y, et al. Liver receptor homolog-1 regulates bile acid homeostasis but is not essential for feedback regulation of bile acid synthesis. Mol Endocrinol 2008; 22: 1345–56.
Inoue Y, Yu AM, Yim SH, Ma X, Krausz KW, Inoue J, et al. Regulation of bile acid biosynthesis by hepatocyte nuclear factor 4alpha. J Lipid Res 2006; 47: 215–27.
Sinal CJ, Tohkin M, Miyata M, Ward JM, Lambert G, Gonzalez FJ . Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell 2000; 102: 731–44.
Kerr TA, Saeki S, Schneider M, Schaefer K, Berdy S, Redder T, et al. Loss of nuclear receptor SHP impairs but does not eliminate negative feedback regulation of bile acid synthesis. Dev Cell 2002; 2: 713–20.
Anakk S, Watanabe M, Ochsner SA, McKenna NJ, Finegold MJ, Moore DD . Combined deletion of Fxr and Shp in mice induces Cyp17a1 and results in juvenile onset cholestasis. J Clin Invest 2011; 121: 86–95.
Buchwald H, Gebhard RL . Localization of bile salt absorption in vivo in the rabbit. Ann Surg 1968; 167: 191–8.
Baker RD, Searle GW . Bile salt absorption at various levels of rat small intenstine. Proc Soc Exp Biol Med 1960; 105: 521–3.
Inagaki T, Choi M, Moschetta A, Peng L, Cummins CL, McDonald JG, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab 2005; 2: 217–25.
Fon Tacer K, Bookout AL, Ding X, Kurosu H, John GB, Wang L, et al. Research resource: Comprehensive expression atlas of the fibroblast growth factor system in adult mouse. Mol Endocrinol 2010; 24: 2050–64.
Potthoff MJ, Boney-Montoya J, Choi M, He T, Sunny NE, Satapati S, et al. FGF15/19 regulates hepatic glucose metabolism by inhibiting the CREB-PGC-1alpha pathway. Cell Metab 2011; 13: 729–38.
Kir S, Beddow SA, Samuel VT, Miller P, Previs SF, Suino-Powell K, et al. FGF19 as a postprandial, insulin-independent activator of hepatic protein and glycogen synthesis. Science 2011; 331: 1621–4.
Yu C, Wang F, Kan M, Jin C, Jones RB, Weinstein M, et al. Elevated cholesterol metabolism and bile acid synthesis in mice lacking membrane tyrosine kinase receptor FGFR4. J Biol Chem 2000; 275: 15482–9.
Vergnes L, Lee JM, Chin RG, Auwerx J, Reue K . Diet1 functions in the FGF15/19 enterohepatic signaling axis to modulate bile acid and lipid levels. Cell Metab 2013; 17: 916–28.
Li S, DDF H, Li B, Luo X, Alderson N, Qiao L, et al. Cytoplasmic tyrosine phosphatase Shp2 coordinates hepatic regulation of bile acid and FGF15/19 signaling to repress bile acid synthesis. Cell Metab 2014; 20: 320–32.
Kong B, Wang L, Chiang JY, Zhang Y, Klaassen CD, Guo GL . Mechanism of tissue-specific farnesoid X receptor in suppressing the expression of genes in bile-acid synthesis in mice. Hepatology 2012; 56: 1034–43.
Kemper JK . Regulation of FXR transcriptional activity in health and disease: Emerging roles of FXR cofactors and post-translational modifications. Biochim Biophys Acta 2011; 1812: 842–50.
Schaap FG, Trauner M, Jansen PL . Bile acid receptors as targets for drug development. Nat Rev Gastroenterol Hepatol 2014; 11: 55–67.
Holt JA, Luo G, Billin AN, Bisi J, McNeill YY, Kozarsky KF, et al. Definition of a novel growth factor-dependent signal cascade for the suppression of bile acid biosynthesis. Genes Dev 2003; 17: 1581–91.
Song KH, Li T, Owsley E, Strom S, Chiang JY . Bile acids activate fibroblast growth factor 19 signaling in human hepatocytes to inhibit cholesterol 7alpha-hydroxylase gene expression. Hepatology 2009; 49: 297–305.
Gineste R, Sirvent A, Paumelle R, Helleboid S, Aquilina A, Darteil R, et al. Phosphorylation of farnesoid X receptor by protein kinase C promotes its transcriptional activity. Mol Endocrinol 2008; 22: 2433–47.
Stravitz RT, Rao YP, Vlahcevic ZR, Gurley EC, Jarvis WD, Hylemon PB . Hepatocellular protein kinase C activation by bile acids: implications for regulation of cholesterol 7 alpha-hydroxylase. Am J Physiol 1996; 271: G293–303.
Rao YP, Stravitz RT, Vlahcevic ZR, Gurley EC, Sando JJ, Hylemon PB . Activation of protein kinase C alpha and delta by bile acids: correlation with bile acid structure and diacylglycerol formation. J Lipid Res 1997; 38: 2446–54.
Le M, Krilov L, Meng JP, Chapin-Kennedy K, Ceryak S, Bouscarel B . Bile acids stimulate PKC alpha autophosphorylation and activation: role in the attenuation of prostaglandin E-1-induced cAMP production in human dermal fibroblasts. Am J Physiol Gastrointest Liver Physiol 2006; 291: G275–87.
Frankenberg T, Miloh T, Chen FY, Ananthanarayanan M, Sun AQ, Balasubramaniyan N, et al. The membrane protein ATPase class I type 8B member 1 signals through protein kinase C zeta to activate the farnesoid X receptor. Hepatology 2008; 48: 1896–905.
Seok S, Kanamaluru D, Xiao Z, Ryerson D, Choi SE, Suino-Powell K, et al. Bile acid signal-induced phosphorylation of small heterodimer partner by protein kinase Czeta is critical for epigenomic regulation of liver metabolic genes. J Biol Chem 2013; 288: 23252–63.
Fu D, Wakabayashi Y, Lippincott-Schwartz J, Arias IM . Bile acid stimulates hepatocyte polarization through a cAMP-Epac-MEK-LKB1-AMPK pathway. Proc Natl Acad Sci U S A 2011; 108: 1403–8.
Woudenberg-Vrenken TE, Conde de la Rosa L, Buist-Homan M, Faber KN, Moshage H . Metformin protects rat hepatocytes against bile acid-induced apoptosis. PLoS One 2013; 8: e71773.
Lien F, Berthier A, Bouchaert E, Gheeraert C, Alexandre J, Porez G, et al. Metformin interferes with bile acid homeostasis through AMPK-FXR crosstalk. J Clin Invest 2014; 124: 1037–51.
Ma K, Xiao R, Tseng HT, Shan L, Fu L, Moore DD . Circadian dysregulation disrupts bile acid homeostasis. PLoS One 2009; 4: e6843.
Li T, Francl JM, Boehme S, Ochoa A, Zhang Y, Klaassen CD, et al. Glucose and insulin induction of bile acid synthesis: mechanisms and implication in diabetes and obesity. J Biol Chem 2012; 287: 1861–73.
Miao J, Xiao Z, Kanamaluru D, Min G, Yau PM, Veenstra TD, et al. Bile acid signaling pathways increase stability of small heterodimer partner (SHP) by inhibiting ubiquitin-proteasomal degradation. Genes Dev 2009; 23: 986–96.
Wang Z, Salih E, Burke PA . Quantitative analysis of cytokine-induced hepatocyte nuclear factor-4alpha phosphorylation by mass spectrometry. Biochemistry 2011; 50: 5292–300.
Yokoyama A, Katsura S, Ito R, Hashiba W, Sekine H, Fujiki R, et al. Multiple post-translational modifications in hepatocyte nuclear factor 4alpha. Biochem Biophys Res Commun 2011; 410: 749–53.
Song KH, Chiang JY . Glucagon and cAMP inhibit cholesterol 7alpha-hydroxylase (CYP7A1) gene expression in human hepatocytes: discordant regulation of bile acid synthesis and gluconeogenesis. Hepatology 2006; 43: 117–25.
Hong YH, Varanasi US, Yang W, Leff T . AMP-activated protein kinase regulates HNF4alpha transcriptional activity by inhibiting dimer formation and decreasing protein stability. J Biol Chem 2003; 278: 27495–501.
Guo H, Gao C, Mi Z, Wai PY, Kuo PC . Phosphorylation of Ser158 regulates inflammatory redox-dependent hepatocyte nuclear factor-4alpha transcriptional activity. Biochem J 2006; 394: 379–87.
Chandra V, Holla P, Ghosh D, Chakrabarti D, Padigaru M, Jameel S . The hepatitis E virus ORF3 protein regulates the expression of liver-specific genes by modulating localization of hepatocyte nuclear factor 4. PLoS One 2011; 6: e22412.
Chellappa K, Jankova L, Schnabl JM, Pan S, Brelivet Y, Fung CL, et al. Src tyrosine kinase phosphorylation of nuclear receptor HNF4alpha correlates with isoform-specific loss of HNF4alpha in human colon cancer. Proc Natl Acad Sci U S A 2012; 109: 2302–7.
Reddy S, Yang W, Taylor DG, Shen X, Oxender D, Kust G, et al. Mitogen-activated protein kinase regulates transcription of the ApoCIII gene. Involvement of the orphan nuclear receptor HNF4. J Biol Chem 1999; 274: 33050–6.
Sun K, Montana V, Chellappa K, Brelivet Y, Moras D, Maeda Y, et al. Phosphorylation of a conserved serine in the deoxyribonucleic acid binding domain of nuclear receptors alters intracellular localization. Mol Endocrinol 2007; 21: 1297–311.
Lee YK, Choi YH, Chua S, Park YJ, Moore DD . Phosphorylation of the hinge domain of the nuclear hormone receptor LRH-1 stimulates transactivation. J Biol Chem 2006; 281: 7850–5.
Acknowledgements
It is gratefully acknowledged that the work in the Gen-Sheng FENG's lab has been supported by the National Institute of Health grants R01HL096125 and R01CA176012 and the American Diabetes Association grant 1-13-BS-048.
The authors have extensively referenced the resources on phosphosite.org, which curates both published and unpublished datasets.
Due to the abundance of review and research papers on BA homeostasis, we regret having to exclude some excellent relevant references due to space limitation.
Author information
Authors and Affiliations
Corresponding author
PowerPoint slides
Rights and permissions
About this article

Cite this article
Li, S., Ni, A. & Feng, Gs. Bridging cell surface receptor with nuclear receptors in control of bile acid homeostasis. Acta Pharmacol Sin 36, 113–118 (2015). https://doi.org/10.1038/aps.2014.118
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2014.118
Keywords
This article is cited by
-
Family reunion of nuclear hormone receptors: structures, diseases, and drug discovery
Acta Pharmacologica Sinica (2015)
