
Abstract
Aim:
To develop a population pharmacokinetic model for the immunosuppressant ciclosporin in Chinese children with aplastic anemia and to identify covariates influencing ciclosporin pharmacokinetics.
Methods:
A total of 102 children with either acquired or congenital aplastic anemia aged 8.8±3.6 years (range 0.9–17.6 years) were included. Therapeutic drug monitoring (TDM) data for ciclosporin were collected. The population pharmacokinetic model of ciclosporin was described using the nonlinear mixed-effects modeling (NONMEM) VI software. The final model was validated using bootstrap and normalized prediction distribution errors.
Results:
A one-compartment model with first-order absorption and elimination was developed. The estimated CL/F was 15.1, which was lower than those of children receiving stem cell or kidney transplant reported in the West (16.9–29.3). The weight normalized CL/F was 0.45 (range: 0.27–0.70) Lh−1·kg−1. The covariate analysis identified body weight, serum creatinine and concomitant administration of the anabolic steroid stanozolol as individual factors influencing the CL/F of ciclosporin.
Conclusion:
Our model could be used to optimize the ciclosporin dosing regimen in Chinese children with aplastic anemia.
Similar content being viewed by others

Introduction
Aplastic anemia is defined as peripheral blood pancytopenia caused by bone marrow failure. Severe aplastic anemia is a life-threatening disorder with an overall mortality of 75%1. The definitive therapy for severe aplastic anemia is hematopoietic stem cell transplantation. Ciclosporin in association with antithymocyte globulin and steroids is an alternative therapeutic option for patients for whom there are no human leukocyte antigen (HLA)-identical related stem cell donors2.
After absorption in the duodenum and the jejunum, ciclosporin binds primarily to lipoproteins and erythrocytes, is widely distributed in the body, and is metabolized by CYP3A4 and CYP3A5 (mainly in the liver and, to a lesser extent, in the intestine and kidneys). The main route of elimination is via the biliary tract3. Ciclosporin has a narrow therapeutic index and high inter-individual pharmacokinetic variability, making individualized therapy essential for treatment success. In current clinical practice, most centers use whole blood trough concentrations (C0) to adjust the individual dose. The primary goal is to maintain C0 within a predefined therapeutic range. Concentration-controlled dosage adaptation is often performed. However, despite widespread use of this strategy, it is still difficult to quickly determine a stable dose that will achieve the target C0 because of numerous demographic, biological and clinical factors influencing ciclosporin pharmacokinetics, including weight, age, serum creatinine and concomitant medication4. Therapeutic drug monitoring (TDM) is mandatory for pediatric patients, who exhibit wider pharmacokinetic variability than adults5.
Population pharmacokinetics, which integrates different effects of variables on drug exposure, has great potential for dosage individualization6. The population pharmacokinetic approach has been used for determining the ciclosporin dose in children who have received a transplant7,8,9. However, pharmacokinetic data for children with aplastic anemia are limited, and a population pharmacokinetic analysis of ciclosporin has not been conducted in this critical population. A thorough understanding of the developmental aspects (ie, the impact of age on ciclosporin clearance) and quantification of the influences of different clinical and biological covariates are required to individualize ciclosporin therapy.
The aims of our study were to develop a population pharmacokinetic model of ciclosporin, using a large TDM database, in children with aplastic anemia and to identify the covariates influencing ciclosporin pharmacokinetics.
Patients and methods
Patients
Children with either acquired or congenital aplastic anemia were considered for inclusion in this observational population pharmacokinetic study if at least one ciclosporin blood concentration was assayed for TDM. The study was undertaken in the Children's Hospital of Zhejiang University, Hangzhou, China.
Ciclosporin (gelatin capsule or liquid solution, NEOCYSPIN, microemulsified formulation, Hangzhou, China) was given orally twice daily with an initial dose of 2.5 mg/kg. Dosage adjustment was based on TDM results to achieve a target C0 of 200 ng/mL10. The concentration of ciclosporin was determined after at least 10 days of consistent administration in order to reach the steady state.
The following data were collected: detailed dosing history, age, gender, body weight, type of disease, white blood cell count, absolute neutrophil count, hemoglobin level, platelet count, serum creatinine concentration and concomitant medications. Patients with incomplete information and patients who did not comply with treatment were excluded. This clinical trial was designed in accordance with legal requirements and the Declaration of Helsinki and was approved by the local ethics committee.
Assay of ciclosporin
Ciclosporin blood concentrations were determined by fluorescence polarization immunoassay technology FPIA (TDX/FLX; Abbott Laboratories, Abbott Park, IL, USA). The lower limit of quantification was 25 ng/mL, and the linearity ranged from 25 to 1500 ng/mL, with a coefficient variation of less than 4%.
Pharmacokinetic analysis
Pharmacokinetic analysis was carried out using the nonlinear mixed effects modeling program NONMEM VI (V2.0; Icon Development Solutions, USA). The first-order conditional estimation (FOCE) method with the interaction option was used to estimate pharmacokinetic parameters and associated variability.
The one-compartment open model with first-order elimination was tested. Inter-individual variability of the pharmacokinetic parameters was estimated using an exponential model expressed as follows:

where θ is the typical population value of the pharmacokinetic parameter and ηiθ is the difference between the log-transformed individual-specific parameter and the log-transformed parameter for the typical individual and represents the random effect for parameter θ in patient i. The ηiθ values are independent, identically distributed random variables that are normally distributed around 0 with variance ω2, and i is the variable for the ith individual.
Residual variability (additive, proportional, exponential or mixed) models were selected according to improvement of the objective function value (OFV) and visual inspection of routine diagnostic plots, including observed (DV) versus individual prediction (IPRED), DV versus population prediction (PRED) and conditional weighted residuals (CWRES) versus PRED.
Covariate analysis
The relationship between body weight and pharmacokinetic parameters was modeled with the following equation:

where Pi is the pharmacokinetic parameter of the ith individual, WTi is the body weight of the ith individual and Pstd is the parameter of an individual with a body weight WTstd of 70 kg. The PWR is the allometric coefficient: 0.75 for the apparent clearance (CL/F) and 1 for the apparent volume of distribution (V/F)11,12.
The additional effects of age, gender, white blood cell count, absolute neutrophil count, hemoglobin level, platelet count, serum creatinine concentration and concomitant medication (prednisone and stanozolol) were investigated as potential variables influencing pharmacokinetic parameters.
The selection of variables was determined using a forward and backward selection process. The OFV determined by NONMEM was used as a diagnostic criterion, with a ΔOFV of 3.84 points corresponding to a statistically significant difference between hierarchical models (P=0.05, χ2 distribution with one degree of freedom). The contribution of each covariate was confirmed by a stepwise backward deletion (ΔOFV=6.64, P=0.01, χ2 distribution).
Model validation
The stability and performance of the final model were assessed by means of an internal validation method involving a nonparametric bootstrap with re-sampling and replacement. Re-sampling was repeated 500 times, and the values of the estimated parameters from the bootstrap procedure were compared with those estimated from the original data set. The entire procedure was performed in an automated fashion, using Perl-speaks-NONMEM (PsN)13.
Simulation-based diagnostics were performed by using normalized prediction distribution errors (NPDE)14. The dataset was simulated 500 times using the final population pharmacokinetic parameters (typical PK parameters, inter- and intra-individual variability and residual error). For NPDE, a cumulative distribution was assembled for each observation with the 500 simulated concentrations. The NPDE is expected to follow the N(0,1) distribution15. The following graphs were plotted using an NPDE R package (v1.2): (i) QQ-plot of the distribution of the NPDE versus the theoretical N(0,1) distribution; (ii) histogram of the NPDE.
Predictive performance
The predictive performance of the developed model was evaluated using the following simulation scenario16:
Ten percent of children (n=10) were randomly removed from the dataset. The parameters for the remaining children (n=92) were re-estimated. The new model was employed to predict the C0 of the 10 children who were not used for the re-estimation of the parameters. An NPDE was assembled for each observation with the 500 simulated concentrations.
Results
One hundred and two children with aplastic anemia were recruited for this population pharmacokinetic study between 2003 and 2009. The mean±SD age was 8.8±3.6 years (range 0.9–17.6), and the mean±SD body weight was 31.3±12.7 kg (range 6.5–69.0). The children's characteristics are shown in Table 1.
Population pharmacokinetic modeling
A total of 592 trough blood ciclosporin concentrations were available for population modeling (a mean of 6 samples per patient). The median (range) of TDM sampling time was 12.1 (10.0–16.0) h post-dose. Inter-individual variability was best described by an exponential model and was then estimated for the absorption rate constant (Ka) and apparent oral clearance (CL/F). The typical Ka value of 0.68 h−1 was fixed according the previously reported value6. Adding a lag time did not improve the model's performance, as judged by the OFV and routine goodness-of-fit plots. Residual variability was best described by a combined proportional and additive model.
After incorporating body weight into the model structure using allometric scaling factors (0.75 for CL/F and 1 for V/F), the OFV significantly decreased, by 15.9. In forward selection processes, influence of body weight, serum creatinine, stanozolol, prednisone and absolute neutrophil count on the CL/F produced significant decreases in the OFV. In the backward selection process, only the significant covariates of weight, serum creatinine and stanozolol were retained (Table 2).
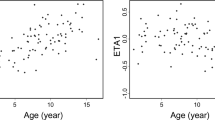
The weight-normalized apparent clearance (CL/F) was 0.45 (range: 0.27–0.70) L·h−1·kg−1. Figure 1 shows the relationship between age and body weight on the allometric weight-normalized CL/F. After incorporation of body weight with allometric scaling, there was no effect of age on the CL/F.

Individual ciclosporin clearance (normalized to weight or allometric weight) plotted against age.
Routine diagnostic plots, including observed versus individual prediction, observed versus population prediction and conditional weighted residuals versus population prediction, indicate goodness of fit (Figure 2). The mean parameter estimates resulting from the bootstrap procedure very closely agreed with their respective values from the final population model, indicating that the estimates for the population pharmacokinetic parameters in the final model were accurate and that the model was stable. The results of 500 bootstrap replicates are summarized in Table 3. The NPDE is presented in Figure 3. The Shapiro-Wilk test of normality was significant, but significance is often observed when analyzing a large number of observations17. Apart from a few outliers, the NPDE plots did not pinpoint a particular deficiency in the model. The good predictive performance of the developed model in predicting C0 is shown in the simulation scenario described above. The mean and variance of the NPDE were -0.19 (Wilcoxon signed rank test, P=0.08) and 1.05 (Fisher variance test, P=0.75), respectively. The NPDE is normally distributed (Shapiro-Wilk test, P=0.22) and is presented in Figure 4. The individual predicted concentrations are significantly correlated with observed concentrations, with a correlation coefficient of 0.796 (P<0.01, Pearson test). The difference is not significant between the two groups (P=0.627, paired t test).

Diagnostic goodness-of-fit plots, including observed (DV) versus individual prediction (IPRED), DV versus population prediction (PRED) and conditional weighted residuals (CWRES) versus PRED.

Normalized prediction distribution errors (NPDE) analysis of the final model. NPDE: QQ-plot of the distribution of the NPDE versus the theoretical N(0, 1) distribution. Histogram of the distribution of the NPDE, with the density of the standard Gaussian distribution overlaid.

Normalized prediction distribution error (NPDE) analysis of the developed model in 10 patients. NPDE: QQ-plot of the distribution of the NPDE versus the theoretical N(0,1) distribution. Histogram of the distribution of the NPDE, with the density of the standard Gaussian distribution overlaid.
Discussion
This first population pharmacokinetic study of ciclosporin in children with aplastic anemia was undertaken to describe the pharmacokinetics of ciclosporin in this special population and to evaluate the influence of developmental, biological and clinical factors affecting drug exposure. Our results showed that a one-compartment model with first-order absorption and elimination was adequate for data modeling and that body weight, serum creatinine concentration and stanozolol administration had significant impacts on CL/F.
The estimated CL/F in children with aplastic anemia was comparable with that reported in children before renal transplantation but seems to be lower than that reported in children after stem cell or renal transplantation (Table 3).
In accord with the principles of allometry and the approach adopted by Fanta et al in studying pediatric renal transplant recipients7, we developed an allometry-based population pharmacokinetic model for ciclosporin: body weight was incorporated with allometric coefficients of 0.75 for CL/F and 1 for V/F11. The allometric approach was confirmed by estimating the CL/F of a patient with a 70 kg standard body weight: the estimated CL/F of 31.5 L/h was in agreement with the published CL/F values for adults18,19. After incorporating body weight into the model, age did not significantly influence the CL/F, which was in agreement with previous results in pediatric renal (pre-) transplant patients7,8.
The effect of renal function on ciclosporin CL/F is controversial. No significant effect was revealed in children after renal and stem cell transplantation8,9. In contrast, Fanta et al reported a significant effect of serum creatinine on CL/F in renal pre-transplant children7. It has been demonstrated that renal dysfunction decreases hepatic drug-metabolism enzyme activities, including CYP3A4, in patients with kidney disease20,21. The correlation between renal function and CYP3A4 activity remains to be confirmed in patients undergoing ciclosporin therapy.
Children undergoing immunosuppressive therapy often take steroids22,23. In vitro studies have shown that steroids are substrates, inducers and inhibitors of CYP3A. Steroids reduce the CYP450 3A metabolism of ciclosporin via competitive inhibition24. Numerous in vivo studies have examined the effects of steroids (methylprednisolone, prednisolone, or prednisone) on ciclosporin concentrations in adult patients; however, controversial results have been reported25. In the present study, 6 different steroids (prednisone, stanozolol, triamcinolone, testosterone undecanoate, methylprednisolone and dexamethasone) were co-administered with ciclosporin, but only prednisone and stanozolol were tested in the model, as there were sufficient samples to test them. Prednisone and stanozolol showed significant effects in the forward covariate selection process, while only stanozolol was retained after backward selection, indicating that stanozolol significantly inhibited CYP3A4-mediated ciclosporin metabolism in children. Careful monitoring of ciclosporin concentrations and dosage adjustment based on TDM results is mandatory when ciclosporin and stanozolol are coadministered.
Our study has some limitations. We planned to prospectively collect a large ciclosporin TDM dataset in children with aplastic anemia. When the recruitment began in 2003, some covariates (eg, cholesterol levels, hematocrit, CYP3A4, and ABCB1 genotype) were not routinely collected, so their impact could not be tested26. In clinical practice, only C0 is routinely measured to adjust the dose of ciclosporin in children with aplastic anemia, and it was thus used in this population pharmacokinetic analysis. In transplanted children, concentrations measured at 2 h post-dose (C2) are better correlated with the AUC5, which is considered to be the best marker of drug exposure to predict the drug's therapeutic effect. Given the important difference in pharmacokinetic parameters between two populations, a prospective study is warranted to evaluate the potential benefits of and/or AUC-based dosing individualization of ciclosporin in children with aplastic anemia.
In conclusion, a population pharmacokinetic model of ciclosporin was developed in Chinese children with aplastic anemia. Weight, serum creatinine and stanozolol administration significantly influenced the ciclosporin CL/F. The model developed here will aid in optimizing ciclosporin doses in children.
Author contribution
Shao-qing NI, Su ZENG, and Zheng-yan ZHAO designed the protocol. Shao-qing NI, Jue WANG, and Shu-qing CHEN participated in protocol development, patient enrolment and data collection. Wei ZHAO and Evelyne Jacqz-Aigrain performed the data analysis. Shao-qing NI and Wei ZHAO prepared the manuscript, which was critically reviewed by the other authors.
References
Li FP, Alter BP, Nathan DG . The mortality of acquired aplastic anemia in children. Blood 1972; 40: 153–62.
Maschan A, Bogatcheva N, Kryjanovskii O, Shneider M, Litvinov D, Mitiushkina T, et al. Results at a single centre of immunosuppression with cyclosporine A in 66 children with aplastic anaemia. Br J Haematol 1999; 106: 967–70.
Yee GC . Recent advances in cyclosporine pharmacokinetics. Pharmacotherapy 1991; 11: 130S–134S.
del Mar Fernández De Gatta M, Santos-Buelga D, Domínguez-Gil A, García MJ . Immunosuppressive therapy for paediatric transplant patients: pharmacokinetic considerations. Clin Pharmacokinet 2002; 41: 115–35.
Zhao W, Fakhoury M, Jacqz-Aigrain E . Developmental pharmacogenetics of immunosuppressants in pediatric organ transplantation. Ther Drug Monit 2010; 32: 688–99.
Zheng QS, Li LJ . Pharmacometrics: a quantitative tool of pharmacological research. Acta Pharmacol Sin 2012; 33: 1337–8.
Fanta S, Jönsson S, Backman JT, Karlsson MO, Hoppu K . Developmental pharmacokinetics of ciclosporin — a population pharmacokinetic study in paediatric renal transplant candidates. Br J Clin Pharmacol 2007; 64: 772–84.
Irtan S, Saint-Marcoux F, Rousseau A, Zhang D, Leroy V, Marquet P, et al. Population pharmacokinetics and bayesian estimator of cyclosporine in pediatric renal transplant patients. Ther Drug Monit 2007; 29: 96–102.
Willemze AJ, Cremers SC, Schoemaker RC, Lankester AC, den Hartigh J, Burggraaf J, et al. Ciclosporin kinetics in children after stem cell transplantation. Br J Clin Pharmacol 2008; 66: 539–45.
Bing H, Siyi Y, Wei Z, Jian L, Minghui D, Li J, et al. The use of anti-human T lymphocyte porcine immunoglobulin and cyclosporine a to treat patients with acquired severe aplastic anemia. Acta Haematol 2010; 124: 245–50.
Anderson BJ, Holford NH . Mechanism-based concepts of size and maturity in pharmacokinetics. Annu Rev Pharmacol Toxicol 2008; 48: 303–32.
Zhao W, Elie V, Roussey G, Brochard K, Niaudet P, Leroy V, et al. Population pharmacokinetics and pharmacogenetics of tacrolimus in de novo pediatric kidney transplant recipients. Clin Pharmacol Ther 2009; 86: 609–18.
Lindbom L, Ribbing J, Jonsson EN . Perl-speaks-NONMEM (PsN) — a Perl module for NONMEM related programming. Comput Methods Programs Biomed 2004; 75: 85–94.
Brendel K, Comets E, Laffont C, Laveille C, Mentré F . Metrics for external model evaluation with an application to the population pharmacokinetics of gliclazide. Pharm Res 2006; 23: 2036–49.
Comets E, Brendel K, Mentré F . Computing normalised prediction distribution errors to evaluate nonlinear mixed-effect models: the npde add-on package for R. Comput Methods Programs Biomed 2008; 90: 154–66.
Zhao W, Piana C, Danhof M, Burger D, Pasqua OD, Jacqz-Aigrain E . Population pharmacokinetics of abacavir in infants, toddlers and children. Br J Clin Pharmacol 2012. DOI: 10.1111/bcp.12024.
Blasco H, Senecal D, Le Gouge A, Pinard E, Benz-de Bretagne I, Colombat P, et al. Influence of methotrexate exposure on outcome in patients treated with MBVP chemotherapy for primary central nervous system lymphoma. Br J Clin Pharmacol 2010; 70: 367–75.
Dunn CJ, Wagstaff AJ, Perry CM, Plosker GL, Goa KL . Cyclosporin: an updated review of the pharmacokinetic properties, clinical efficacy and tolerability of a microemulsion-based formulation (neoral) 1 in organ transplantation. Drugs 2001; 61: 1957–2016.
Saint-Marcoux F, Marquet P, Jacqz-Aigrain E, Bernard N, Thiry P, Le Meur Y, et al. Patient characteristics influencing ciclosporin pharmacokinetics and accurate Bayesian estimation of ciclosporin exposure in heart, lung and kidney transplant patients. Clin Pharmacokinet 2006; 45: 905–22.
Nolin TD, Naud J, Leblond FA, Pichette V . Emerging evidence of the impact of kidney disease on drug metabolism and transport. Clin Pharmacol Ther 2008; 83: 898–903.
Nolin TD, Appiah K, Kendrick SA, Le P, McMonagle E, Himmelfarb J . Hemodialysis acutely improves hepatic CYP3A4 metabolic activity. J Am Soc Nephrol 2006; 17: 2363–7.
Benfield MR, Stablein D, Tejani A . Trends in immunosuppressive therapy: a report of the north American pediatric renal transplant cooperative study (NAPRTCS). Pediatr Transplant 1999; 3: 27–32.
Ettenger RB . New immunosuppressive agents in pediatric renal transplantation. Transplant Proc 1998; 30: 1956–8.
Nakamura H, Nakasa H, Ishii I, Ariyoshi N, Igarashi T, Ohmori S, et al. Effects of endogenous steroids on CYP3A4-mediated drug metabolism by human liver microsomes. Drug Metab Dispos 2002; 30: 534–40.
Lam S, Partovi N, Ting LS, Ensom MH . Corticosteroid interactions with cyclosporine, tacrolimus, mycophenolate, and sirolimus: fact or fiction? Ann Pharmacother 2008; 42: 1037–47.
Zhou H, Gao Y, Cheng XL, Li ZD . Population pharmacokinetics of cyclosporine A based on NONMEM in Chinese allogeneic hematopoietic stem cell transplantation recipients. Eur J Drug Metab Pharmacokinet 2012; 37: 271–8.
Acknowledgements
We thank Dingtai Ren for his great support in recording information and sample treatments and David Shen for his kindly help in our population pharmacokinetic experiments. We also sincerely thank all the participating patients and their families for their cooperation.
This work was supported by grants from the Natural Science Foundation of Zhejiang Province (Y2080183), the National Natural Science Foundation of China (81273607), the Science and Technologies Foundation of China (2009ZX09304) and the National Science and Technology Major Projects for “Major New Drugs Innovation and Development” (2013ZX09303003). Doctor Wei Zhao and Professor Evelyne Jacqz-Aigrain received support from the European commission for their pediatric pharmacology collaborative work in China (FP7 Global research in pediatrics GRIP, grant agreement No 261060).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Ni, Sq., Zhao, W., Wang, J. et al. Population pharmacokinetics of ciclosporin in Chinese children with aplastic anemia: effects of weight, renal function and stanozolol administration. Acta Pharmacol Sin 34, 969–975 (2013). https://doi.org/10.1038/aps.2013.9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2013.9
Keywords
This article is cited by
-
Population pharmacokinetics of cyclosporine in Chinese children receiving hematopoietic stem cell transplantation
Acta Pharmacologica Sinica (2019)
