
Abstract
Aim:
To study the conformational changes of Aβ42 and discover novel inhibitors of both Aβ42 aggregation and β-secretase (BACE1).
Methods:
A molecular dynamics (MD) simulation at a microsecond level was performed to explore stable conformations of Aβ42 monomer in aqueous solution. Subsequently, structure-based virtual screening was used to search for inhibitors of both Aβ42 aggregation and BACE1. Protein purification and in vitro activity assays were performed to validate the inhibition of the compounds identified via virtual screening.
Results:
The initial α-helical conformation of Aβ42, which was unstable in aqueous solution, turned into a β-sheet mixed with a coil structure through a transient and fully random coil. The conformation of Aβ42 mainly comprising β-sheets and coils structure was used for further virtual screening. Five compounds were identified as inhibitors for Aβ42 aggregation, and one of them, AE-848, was discovered to be a dual inhibitor of both Aβ42 aggregation and BACE1, with IC50 values of 36.95 μmol/L and 22.70 μmol/L, respectively.
Conclusion:
A helical to β-sheet conformational change in Aβ42 occurred in a 1.8 microsecond MD simulation. The resulting β-sheet structure of the peptide is an appropriate conformation for the virtual screening of inhibitors against Aβ42 aggregation. Five compounds were identified as inhibitors of Aβ42 aggregation by in vitro activity assays. It was particularly interesting to discover a dual inhibitor that targets both Aβ42 aggregation and BACE1, the two crucial players in the pathogenesis of Alzheimer's disease.
Similar content being viewed by others

Introduction
Alzheimer's disease (AD), a neurodegenerative disorder, is pathologically characterized by the presence of insoluble amyloid plaques and neurofibrillary tangles in the brain1. The major components of the plaques are amyloid β-peptides (Aβs) consisting of 39–43 residues, which arise from the consecutive cleavage of the amyloid precursor protein (APP) by β- and γ-secretase, respectively2,3,4. The amyloid hypothesis suggests that misfolding of Aβ leads to its dysfunction and aggregation; the latter is associated with a cascade of neuropathogenetic events to produce the cognitive and behavioral decline that are hallmarks of AD5,6. The peptides containing 40 (Aβ40) and 42 (Aβ42) residues are the two predominant isoforms of Aβ3,7,8. Despite the low levels of Aβ42 produced under physiological conditions (approximately 10% of total Aβs), Aβ42 has a higher propensity for aggregation and displays an enhanced neurotoxicity compared to Aβ409.
In the production pathway of Aβs, β-secretase (β-site amyloid precursor protein-cleaving enzyme, BACE1) initiates the cleavage of APP to form a soluble N-terminal ectodomain together with a membrane-bound 99-residue C-terminal fragment (CTF99)10, which is the first step of amyloidogenic APP metabolism. CTF99 is then further processed by γ-secretase to produce Aβ as well as the APP intracellular domain. Because BACE1 is the rate-limiting step in the production of Aβs11, it represents a prime target for the development of inhibitors that may serve as drugs in the treatment and/or prevention of AD.
In the past decades, many X-ray structures of the apo BACE1 and the BACE1-inhibitor complex have been determined, which provide detailed information about the structure and functional analysis of BACE111,12,13,14,15,16. BACE1 is a membrane-anchored aspartic protease containing three distinct domains: an N-terminal ectodomain, a single transmembrane domain, and a cytosolic C-terminus. The ectodomain is the protease domain and has the correct topological orientation for cleavage of APP at sites susceptible to BACE111.
In the current study, a 1.8-μs long molecular dynamics (MD) simulation was performed to obtain an appropriate Aβ42 monomer conformation in aqueous solution for a virtual screening. A relatively stable conformation containing a long β-sheet and coil was observed in the trajectory. A structure-based virtual screening was performed based on this Aβ42 conformation, and five compounds were eventually identified as inhibitors of Aβ42 aggregations, one of which also exhibited inhibitory activity against BACE1. These observations contributed to our understanding of the mechanism underlying amyloid formation and may be useful in the design of novel compounds that target both Aβ generation and aggregation to prevent neurotoxicity induced by Aβs.
Materials and methods
MD simulation of Aβ42
Initial coordinates of the Aβ40 monomer were directly extracted from the nuclear magnetic resonance(NMR) structure determined in aqueous SDS micelles at pH 5.1 (PDB entry 1ba4, state 2)17. Two additional residues were added at the C-terminus of Aβ40 to generate Aβ42. The protocol for building the simulation boxes and the parameter settings used for the MD run were similar to our earlier study18. The peptide was first placed in a suitably sized box where the minimum distance from the atoms of the peptide to the box wall was 2.0 nm. The box was solvated with simple point charge (SPC) water, and the peptide-water system was subjected to energy minimization. Afterward, counter ions were added to the system to provide a neutral simulation system. The resulting system was subjected to a second energy minimization to remove unfavorable contacts and equilibrated for 500 ps with positional restraints on the peptide atoms. Finally, the entire system was simulated for 1.8-μs without any restraints.
MD simulations were performed with the GROMACS 4.5.3 software package19 using the constant number, pressure, and temperature (NPT) method with periodic boundary conditions. The GROMOS53a6 force field20 was applied to the peptide. Analyses were performed with the GROMACS package based on the trajectory at 100 ps intervals. Secondary structure analyses were performed using the defined secondary structure of proteins (DSSP) method21. Graphics were generated with the Pymol (DeLano, WL, The PyMOL Molecular Graphics System, 2002, http://www.pymol.org) and Origin (version 8E; Microcal Software Inc, Northampton, MA) software packages. A clustering method (g_cluster tools in Gromacs package) was used to classify the representative conformations of Aβ42 in the MD trajectory. The cluster cutoff used ensured that the root mean square deviation (RMSD) of any two snapshots included in a single cluster was less than 0.25 nm.
Virtual screening
A snapshot of Aβ42 extracted from the MD trajectory at 1.8 μs, which represents a relatively stable conformation of Aβ42, was used for virtual screening. Approximately 200 000 compounds in the SPECS chemical database were screened. In the preparation of Aβ42, hydrogen atoms and charges were added via the Protein Preparation module of the Maestro software package (Schrodinger, Inc) using the “Preparation and refinement” option. The restrained partial minimization was terminated when the RMSD reached a maximum value of 0.3 Å. A grid-enclosing box was placed on the center of Aβ42 to include residues located within 10 Å of the center. A scaling factor of 1.0 Å was set for van der Waals (VDM) radii of the receptor atoms. For the docking process, the Dock 4.0 software package22 was used to prescreen all compounds, followed by accurate docking using Glide calculations with the Maestro v7.5 software package. Standard precision (SP) docking was adopted to generate the minimized pose, and the Glide scoring function (G-Score) was used to select the final top 2000 compounds.
The coordinates of BACE1 were directly extracted from the PDB database (PDB code: 3tpp)11 with the ligand and water molecules removed. The same protein preparation and docking process used for Aβ42 was used for BACE1 except the box was centered on the position of the ligand. Only the dual inhibitor was docked to BACE1 using Glide.
Protein purification and in vitro assay
The Aβ42 peptide was purchased from Ziyu Biotechnology Co Ltd (Shanghai). A detailed description of the production of recombinant human BACE1 was described in our previous publication11. Briefly, BACE1 proteins containing residues 43–454 were expressed in E. coli as inclusion bodies, which were then denatured and refolded into the active monomer.
A stock solution of Aβ42 was prepared according to the following protocol. Aβ42 was dissolved in DMSO to reach a concentration of 5 mg/mL (1.15 mmol/L) and Thioflavin T was dissolved in distilled water to reach a final concentration of 1 mmol/L. These stock solutions were stored at -20 °C. For each compound, 2 μL of its stock solution (1 mmol/L in DMSO), 0.5 μL Thioflavin T, and 1 μL of the Aβ42 stock solution were added sequentially, which were then diluted with 36.5 μL of a phosphate-buffered saline (PBS) solution (50 mmol/L of Na2HPO4 and 100 mmol/L of NaCl, pH 7.4) to reach a final volume of 40 μL. The final DMSO concentration in the 40-μL reaction volume was kept at less than 10%. The samples were covered with aluminum foil and incubated at 37 °C overnight.
The BACE1 inhibitory activity assay kit was purchased from Invitrogen (Carlsbad, CA, USA). The assay was performed according to the manufacturer's protocol. The enzyme, substrate, and compounds were diluted in a reaction buffer (50 mmol/L sodium acetate, pH 4.5) to make 3×working solutions. The assay was performed in a black 384-well microplate with a final volume of 30 μL per well, which contained 10 μL of 3×substrate, enzyme, and compound stocks, respectively. The final concentration of DMSO was less than 3% (v/v). The reaction mixture was incubated at 37 °C for 90 min with oscillations. Next, 10 μL of a stop solution (2.5 mol/L sodium acetate) was added to stop the reaction. Finally, the fluorescent intensity of the enzymatic product was measured at Ex/Em=535 nm/585 nm on a BioTek SYNERGY4 reader (BioTek, Winooski, USA).
Results
Conformational conversion of Aβ42 from an α-helix to β-sheets
As indicated by many NMR structures23,24,25,26, the α-helical structure of Aβ42 is unstable in aqueous solution at a neutral pH. A 1.8-μs simulation trajectory was, therefore, performed to explore the dynamic conformations of full-length Aβ42 in aqueous solution. The secondary structure analyses of the peptide versus the simulation time, together with the snapshots of Aβ42 extracted along the trajectory, are shown in Figure 1. The initial long α-helical structure of the peptide disappeared after several nanoseconds and short β-sheets appeared at the C-terminus of the peptide, which is in agreement with our earlier observation18. The short β-sheets were maintained for less than 100 ns, and a coiled structure conformation appeared approximately 100 ns (Figure 1).

Conformational conversion of Aβ42 in aqueous solution. (A) Representative snapshots of Aβ42 extracted from the 1.8-μs trajectory. (B) Secondary structures as a function of time for Aβ42 were calculated by the program DSSP. The structures of the peptide were examined at every 100 ps. (C) Time dependence of the Cα RMSD of the peptide with respect to the starting structure (black) and the previous snapshot (red) during the simulation.
The representative snapshot of Aβ42 extracted at 100 ns shows that all the residues of the peptide adopted a random coiled structure at this time point. Only a few nanoseconds later, however, short β-sheets appeared in both the N-terminus and C-terminus of Aβ42. A third short β-strand was formed near residue K28 a few hundred nanoseconds later. After approximately 1.3 μs, long β-sheets began to form mostly at the N-terminus of the peptide. In summary, the 1.8-μs MD trajectory revealed that the full-length Aβ42 underwent a conformational conversion from an initial α-helix to a coil, then to long β-sheets in aqueous solution at a neutral pH. The time-dependent Cα RMSD using the starting conformation or the previous snapshot as a reference shown in Figure 1C further supports the conclusion that large evolutionary conformational changes occurred to the peptide during the simulation.
The percentages of the secondary structures of Aβ42 observed (helix, coil, and β-sheet) were calculated and plotted as a function of the simulation time in Figure 2A. The percentage of the helix dropped off within the first 100 ns, whereas the percentage of the coil and β-sheet increased after 100 ns. At approximately 100 ns, the coil percentage reached a maximum. At this time point, almost all residues adopted coiled structures, and no helices or β-sheets were observed. This finding is consistent with the DSSP plot shown in Figure 1B. From approximately 0.13 to 1.2 μs, the percentages of the coil and β-sheet are relatively flat, with values of approximately 75% and 25%, respectively. During this period, it was primarily the G9-H13, N27-G29, and G38-I41 residues that formed β-strands. After 1.2 μs, the percentage of β-sheets began to increase and reached a maximum value approximately 1.4 μs. Conversely, residues with coiled structures were reduced and the percentage of β-sheets was higher than that of the coil during this period. When the percentage of β-sheets was the highest (from approximately 1.35 to 1.45 μs), long β-strands were formed in residues A2-H6, S8-A21, and N27-I41. From approximately 1.53 to 1.8 μs, approximately 40% and 60% of the residues adopted β-sheet and coiled structures, respectively. Four β-strands in the A2-R5, S8-V17, S28-A30, and G39-V40 residues were maintained. Therefore, a coil-dominated structure mixed with β-sheets is favored by Aβ42 in aqueous solution (Figures 1A and 2D). The potential energy of the peptide also implies that such a mixed structure is an energy-favorable one for Aβ42 because the energy decreased from an initial -1000 kJ/mol to approximately -2000 kJ/mol at the end of the simulation (Figure 2B). The number of main-chain hydrogen bonds of Aβ42 decreased in the first 100 ns, which is in agreement with the secondary structure change from the α-helix to a coil in this time period. Subsequently, the number of hydrogen bonds increased to approximately 11 and was maintained until approximately 1.3 μs. From 1.3 μs until the end of the simulation, approximately 5 additional main-chain hydrogen bonds were formed (Figure 2C).

Percentage of secondary structures (A), potential energy (B), and main-chain hydrogen bonds (C) of Aβ42 in the 1.8-μs trajectory. (D) Cluster analysis of the conformations of the peptide in the trajectory. Cluster centered structures for the top 6 clusters, which contained 43.5%, 24.2%, 14.9%, 13.9%, 1.2%, and 0.5% of total snapshots, respectively. Cluster analysis was performed on all the snapshots extracted from the whole trajectory at every 100 ps.
Clustering analysis was performed based on all snapshots extracted from the whole trajectory every 100 ps. A cutoff of 0.25 nm of the Cα RMSD for any two snapshots included in one cluster was used. 45 clusters were obtained, in which the top six clusters included 98.2% of all snapshots. Cluster-centered structures of the six clusters are shown in Figure 2D. Four of the six centered structures consisted of β-sheets and coils, which occupied 96.5% of all snapshots. Less than 3.5% of the snapshots contain α-helical structures. Therefore, the cluster structure analysis also indicates that (1) the helical structure of Aβ42 was unstable, whereas the β-sheets containing conformations were relatively stable in aqueous solution at a neutral pH; (2) there was a high probability that both the N- and C-termini would form β-sheets; and (3) the long β-sheets formed in residues D7-K16 and S26-I31 were important for stabilizing the whole peptide structure.
Virtual screening and in vitro assay validation
Because the MD simulation revealed that the mixed coil and β-sheet structure is a favored structure for Aβ42 monomer in aqueous solution, a snapshot of the peptide derived from the end of the trajectory was chosen for the structure-based virtual screening of inhibitors (Figure 1A). A schematic representation of the overall approach used to discover inhibitors via virtual screening and in vitro assays is presented in Figure 3A. The entire Aβ42 peptide was taken as the binding pocket used in the virtual screening because the exact binding location of small molecules is unknown. The DOCK program was used for the preliminary screening of compounds included in the SPECS database (approximately 200 000 compounds). The energy score of the Aβ42-compound complex was cut to -22.00 kcal/mol. As a result, the top 29824 compounds were selected for further screening. These compounds were then docked to Aβ42 with the Maestro Glide module using the standard precision (SP) mode. The top 2000 compounds with a Glide score (Gscore) less than -3.75 were selected. Next, scaffold diversity analysis was performed using the cluster molecules component of Pipeline Pilot 7.5 to select the final 183 representative compounds, which were purchased for the in vitro assay tests.

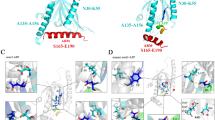
Structure-based virtual screening inhibitors of Aβ42 aggregations and BACE1 based on the compounds from SPECS database. (A) Schematic representation of the overall procedure to discover the inhibitor. (B, C) Ligplot representation of AE-848 interacting with Aβ42 (B) and BACE1 (C).
To test the inhibitory activity of compounds that were selected in the virtual screening, Aβ42 aggregation and BACE1 activity assays were performed. The IC50 values were determined experimentally as described in the Materials and Methods section. Five compounds were found to exhibit inhibitory activities against Aβ42 aggregation. The chemical structures of these compounds are shown in Figure 4A. The IC50 of these compounds are 36.95 (AE-848, Figure 4B), 23.05 (AG-227), 21.59 (AJ-030), 17.41 (AG-690), and 188.56 μmol/L (AA-504), respectively. These compounds were further tested for their inhibitory activities against BACE1. Finally, AE-848 was identified as a dual inhibitor because it inhibits the aggregation of Aβ42 and the catalytic activity of BACE1. The IC50 value of AE-848 against BACE1 was 22.70 μmol/L (Figure 4C).

(A) The chemical structures of 5 inhibitors against Aβ42 aggregation. (B, C) The inhibitory activity profile of AE-848 against Aβ42 aggregation (B) and BACE1 (C).
The interactions between AE-848 and Aβ42 predicted from Glide docking are presented as a ligplot27 (Figure 3B). In this complex, AE-848 bound to an area consisting primarily of the H13, G38, V39, V40, and I41 residues. Hydrogen bonds between AE-848 and the main-chain of G38, V39, and I41 of Aβ42 were formed. A similar protocol and parameter set were used to study the binding mode of AE-848 to BACE1 using Glide. AE-848 docked to the binding site of BACE1 that is surrounded by the D32, G34, Y71, T72, Q73, and D228 residues. The compound interacted with the main-chain of T72 and Q73 as well as the side-chain of D32 through hydrogen bonds (Figure 3C).
Discussion
In the past two decades, multiple technologies such as NMR, electron microscopy (EM), X-ray crystal diffraction, and atomic force microscopy (AFM) have been used to determine the structure of the Aβ monomer and oligomers under various conditions17,28,29,30,31,32,33. However, as a consequence of the Aβ peptide's high propensity for aggregation, it is difficult to solve the structure of the peptide as a monomer or an oligomer at a high resolution. MD simulations of Aβ were, therefore, performed based on the available NMR structures18,34,35,36, which provided intuitionistic dynamic views of Aβ conformational conversion. Because most previous simulation studies have focused on fragments rather than full length Aβ, it is highly likely that key interactions or effects involved in aggregation-associated conformational changes of Aβ have been missed. In the present study, a 1.8-μs MD simulation of Aβ42 was performed in liquid solution at a neutral pH and room temperature, which represents the longest conventional MD simulation used to date to investigate the conformational conversion of full length Aβ42 at the atomic level. Evident conformational conversion was observed and β-sheet rich conformations of Aβ42 existed for over 200 ns. These data suggest that the monomeric Aβ42 may also have a high propensity for β-sheet formation in aqueous solution even though the main secondary structure for the peptide is a coil.
As a consequence of the high aggregation properties of Aβ (and Aβ42 in particular) as well as the cytotoxicity of Aβ oligomers, there is tremendous interest in preventing Aβ aggregation by identifying inhibitors of Aβs37,38,39. Various inhibitors, such as antibodies, small compounds, Chinese herbal medicines, and peptides, have been explored in the prevention of Aβ aggregation37,38,40,41,42. Sievers et al reported inhibitors against the amyloid fibril formation based on a steric zipper structure of an Aβ fragment (VQIVYK) where the D-amino-acid hexapeptide was designed via a computational method to interact with the fragment and to cap its fibril ends37. Kroth et al discovered and designed a series of small compounds to inhibit the fibril formation of Aβs at μmol/L concentrations38,43. In addition, Jan-Philipp Bach and Richard Dodel reviewed the study of naturally occurring autoantibodies against β-amyloid that improve cognition in transgenic mice by interfering with oligomers of Aβ40.
While the five compounds we identified inhibited the aggregation of Aβ42 at μmol/L concentrations, one of the five compounds was found to have dual inhibitory activities against Aβ42 aggregation and BACE1. Moreover, the structure of AE-848 is relatively small and can be used as a fragment for further modification targeting to Aβ42 aggregation or/and BACE1. Most interestingly, these inhibitors were identified via a virtual screening that was based on a specific conformation of the peptide. They may also serve as molecular probes to precisely study the conformational changes associated with Aβ42 aggregation. A further modification based on the identified compound AE-848 is underway to facilitate the identification of additional small molecules that interact with Aβ42 aggregation or BACE1 with a higher binding affinity.
In summary, MD simulation and structure-based virtual screening computational methods were employed to seek out potential inhibitors of Aβ42 aggregation. It was observed that in aqueous solution, the α-helical structure of the Aβ42 monomer was unstable and that the monomeric peptide favors a coil-dominated structure mixed with β-sheets. The virtual screening based on the favored structure of Aβ42 identified 5 compounds that inhibit the aggregation of the Aβ42 peptide at μmol/L concentrations. Secondary inhibitory activity assessments of these compounds against BACE1 identified a dual inhibitor of both Aβ42 aggregation and BACE1 (AE-848). Further structural modifications based on this compound are underway.
Author contribution
Yuan-yuan WANG performed the MD simulation and virtual screening. Li LI, Tian-tian CHEN, and Wu-yan CHEN performed experiments. Yuan-yuan WANG and Ye-chun XU analyzed the data and wrote the paper.
References
Selkoe DJ . The maolecular pathology of Alzheimer's disease. Neuron 1991; 6: 487–98.
Miller DL, Papayannopoulos IA, Styles J, Bobin SA, Lin YY, Biemann K, et al. Peptide compositions of the cerebrovascular and senile plaque core amyloid deposits of Alzheimer's disease. Arch Biochem Biophys 1993; 301: 41–52.
Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, et al. The precursor of Alzheimer's disease amyloid A4 protein resembles a cell-surface receptor. Nature 1987; 325: 733–6.
Mattson MP . Cellular actions of beta-amyloid precursor protein and its soluble and fibrillogenic derivatives. Physiol Rev 1997; 77: 1081–132.
Selkoe DJ . Alzheimer's disease: genes, proteins, and therapy. Physiol Rev 2001; 81: 741–66.
Hardy J, Selkoe DJ . The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 2002; 297: 353–6.
Speretta E, Jahn TR, Tartaglia GG, Favrin G, Barros TP, Imarisio S, et al. Expression in drosophila of tandem amyloid beta peptides provides insights into links between aggregation and neurotoxicity. J Biol Chem 2012; 287: 20748–54.
Xia W . Brain amyloid beta protein and memory disruption in Alzheimer's disease. Neuropsychiatr Dis Treat 2010; 6: 605–11.
Liu D, Xu Y, Feng Y, Liu H, Shen X, Chen K, et al. Inhibitor discovery targeting the intermediate structure of beta-amyloid peptide on the conformational transition pathway: implications in the aggregation mechanism of beta-amyloid peptide. Biochemistry 2006; 45: 10963–72.
Vassar R . BACE1: the beta-secretase enzyme in Alzheimer's disease. J Mol Neurosci 2004; 23: 105–14.
Xu Y, Li MJ, Greenblatt H, Chen W, Paz A, Dym O, et al. Flexibility of the flap in the active site of BACE1 as revealed by crystal structures and molecular dynamics simulations. Acta Crystallogr D Biol Crystallogr 2012; 68: 13–25.
Rueeger H, Lueoend R, Rogel O, Rondeau JM, Mobitz H, Machauer R, et al. Discovery of cyclic sulfone hydroxyethylamines as potent and selective beta-site APP-cleaving enzyme 1 (BACE1) inhibitors: structure-based design and in vivo reduction of amyloid beta-peptides. J Med Chem 2012; 55: 3364–86.
Mandal M, Zhu Z, Cumming JN, Liu X, Strickland C, Mazzola RD, et al. Design and validation of bicyclic iminopyrimidinones as beta amyloid cleaving enzyme-1 (BACE1) inhibitors: conformational constraint to favor a bioactive conformation. J Med Chem 2012; 55: 9331–45.
Zhou P, Li Y, Fan Y, Wang Z, Chopra R, Olland A, et al. Pyridinyl aminohydantoins as small molecule BACE1 inhibitors. Bioorg Med Chem Lett 2010; 20: 2326–9.
Shimizu H, Tosaki A, Kaneko K, Hisano T, Sakurai T, Nukina N . Crystal structure of an active form of BACE1, an enzyme responsible for amyloid beta protein production. Mol Cell Biol 2008; 28: 3663–71.
Malamas MS, Barnes K, Hui Y, Johnson M, Lovering F, Condon J, et al. Novel pyrrolyl 2-aminopyridines as potent and selective human beta-secretase (BACE1) inhibitors. Bioorg Med Chem Lett 2010; 20: 2068–73.
Coles M, Bicknell W, Watson AA, Fairlie DP, Craik DJ . Solution structure of amyloid beta-peptide (1–40) in a water-micelle environment. Is the membrane-spanning domain where we think it is? Biochemistry 1998; 37: 11064–77.
Xu Y, Shen J, Luo X, Zhu W, Chen K, Ma J, et al. Conformational transition of amyloid beta-peptide. Proc Natl Acad Sci U S A 2005; 102: 5403–7.
Hess B, Kutzner C, van der Spoel D, Lindahl E . GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. journal of chemical theory and computation. J Chem Theory Comput 2008; 4: 435–47.
Oostenbrink C, Villa A, Mark AE, van Gunsteren WF . A biomolecular force field based on the free enthalpy of hydration and solvation: the GROMOS force-field parameter sets 53A5 and 53A6. J Comput Chem 2004; 25: 1656–76.
Kabsch W, Sander C . Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983; 22: 2577–637.
Ewing TJ, Makino S, Skillman AG, Kuntz ID . DOCK 4.0: search strategies for automated molecular docking of flexible molecule databases. J Comput Aided Mol Des 2001; 15: 411–28.
Tomaselli S, Esposito V, Vangone P, van Nuland NA, Bonvin AM, Guerrini R, et al. The alpha-to-beta conformational transition of Alzheimer's Abeta-(1–42) peptide in aqueous media is reversible: a step by step conformational analysis suggests the location of beta conformation seeding. Chembiochem 2006; 7: 257–67.
Zhang S, Iwata K, Lachenmann MJ, Peng JW, Li S, Stimson ER, et al. The Alzheimer's peptide a beta adopts a collapsed coil structure in water. J Struct Biol 2000; 130: 130–41.
Stroud JC, Liu C, Teng PK, Eisenberg D . Toxic fibrillar oligomers of amyloid-beta have cross-beta structure. Proc Natl Acad Sci U S A 2012; 109:7717–22.
Ito M, Johansson J, Stromberg R, Nilsson L . Unfolding of the amyloid beta-peptide central helix: mechanistic insights from molecular dynamics simulations. PLoS One 2011; 6: e17587.
Wallace AC, Laskowski RA, Thornton JM . LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng 1995; 8: 127–34.
Vivekanandan S, Brender JR, Lee SY, Ramamoorthy A . A partially folded structure of amyloid-beta(1–40) in an aqueous environment. Biochem Biophys Res Commun 2011; 411: 312–6.
Streltsov VA, Varghese JN, Masters CL, Nuttall SD . Crystal structure of the amyloid-beta p3 fragment provides a model for oligomer formation in Alzheimer's disease. J Neurosci 2011; 31: 1419–26.
Crescenzi O, Tomaselli S, Guerrini R, Salvadori S, D'Ursi AM, Temussi PA, et al. Solution structure of the Alzheimer amyloid beta-peptide (1–42) in an apolar microenvironment. Similarity with a virus fusion domain. Eur J Biochem 2002; 269: 5642–8.
Luhrs T, Ritter C, Adrian M, Riek-Loher D, Bohrmann B, Dobeli H, et al. 3D structure of Alzheimer's amyloid-beta(1–42) fibrils. Proc Natl Acad Sci U S A 2005; 102: 17342–7.
Yu L, Edalji R, Harlan JE, Holzman TF, Lopez AP, Labkovsky B, et al. Structural characterization of a soluble amyloid beta-peptide oligomer. Biochemistry 2009; 48: 1870–7.
Connelly L, Jang H, Arce FT, Ramachandran S, Kagan BL, Nussinov R, et al. Effects of point substitutions on the structure of toxic Alzheimer's beta-amyloid channels: atomic force microscopy and molecular dynamics simulations. Biochemistry 2012; 51: 3031–8.
Reddy G, Straub JE, Thirumalai D . Influence of preformed Asp23-Lys28 salt bridge on the conformational fluctuations of monomers and dimers of Abeta peptides with implications for rates of fibril formation. J Phys Chem B 2009; 113: 1162–72.
Lam AR, Teplow DB, Stanley HE, Urbanc B . Effects of the Arctic (E22→G) mutation on amyloid beta-protein folding: discrete molecular dynamics study. J Am Chem Soc 2008; 130: 17413–22.
Yang M, Teplow DB . Amyloid beta-protein monomer folding: free-energy surfaces reveal alloform-specific differences. J Mol Biol 2008; 384: 450–64.
Sievers SA, Karanicolas J, Chang HW, Zhao A, Jiang L, Zirafi O, et al. Structure-based design of non-natural amino-acid inhibitors of amyloid fibril formation. Nature 2011; 475: 96–100.
Kroth H, Ansaloni A, Varisco Y, Jan A, Sreenivasachary N, Rezaei-Ghaleh N, et al. Discovery and structure activity relationship of small molecule inhibitors of toxic beta-amyloid-42 fibril formation. J Biol Chem 2012; 287: 34786–800.
Hard T, Lendel C . Inhibition of amyloid formation. J Mol Biol 2012; 421: 441–65.
Bach JP, Dodel R . Naturally occurring autoantibodies against beta-Amyloid. Adv Exp Med Biol 2012; 750: 91–9.
Wang Y, Huang LQ, Tang XC, Zhang HY . Retrospect and prospect of active principles from Chinese herbs in the treatment of dementia. Acta pharmacol Sin 2010; 31: 649–64.
Deane R, Singh I, Sagare AP, Bell RD, Ross NT, LaRue B, et al. A multimodal RAGE-specific inhibitor reduces amyloid beta-mediated brain disorder in a mouse model of Alzheimer disease. J Clin Invest 2012; 122: 1377–92.
Ghosh AK, Brindisi M, Tang J . Developing beta-secretase inhibitors for treatment of Alzheimer's disease. J Neurochem 2012; 120: 71–83.
Acknowledgements
This study was supported by the “100 Talents Project” of CAS (to Ye-chun XU), the State Key Program of Basic Research of China (Grant No 2009CB918501), and the National Natural Science Foundation of China (No 91013010 and 21172233). Computational resources were supported by the National Supercomputing Center in Tianjin (Tianhe-1), the Shanghai Supercomputer Center and the Computer Network Information Center of the Chinese Academy of Sciences.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wang, Yy., Li, L., Chen, Tt. et al. Microsecond molecular dynamics simulation of Aβ42 and identification of a novel dual inhibitor of Aβ42 aggregation and BACE1 activity. Acta Pharmacol Sin 34, 1243–1250 (2013). https://doi.org/10.1038/aps.2013.55
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2013.55
Keywords
This article is cited by
-
Conformation and dynamics of the C-terminal region in human phosphoglycerate mutase 1
Acta Pharmacologica Sinica (2017)
