
Abstract
Aim:
To investigate the effects of puerarin (Pue), an isoflavone derived from Kudzu roots, on angiotensin II (Ang II)-induced hypertrophy of cardiomyocytes in vivo and in vitro.
Methods:
C57BL/6J mice were infused with Ang II and treated with Pue (100 mg·kg−1·d−1, po) for 15 d. After the treatment, systolic blood pressure (SBP) and left ventricular wall thickness were assessed. The ratios of heart weight to body weight (HW/BW) and left ventricular weight to body weight (LVW/BW) were determined, and heart morphometry was assessed. Expression of fetal-type genes (ANP, BNP and β-MHC) in left ventricles was measured using semi-quantitative RT-PCR. Mouse primary cardiomyocytes were treated with Pue (50, 100, 200 μmol/L), then exposed to Ang II (1 μmol/L). ROS level was examined with flow cytometry, the binding activity of NF-κB was determined using EMSA. Western blot was used to measure the levels of ERK1/2, p38 and NF-κB pathway proteins. [3H]leucine incorporation was used to measure the rate of protein synthesis.
Results:
Oral administration of Pue significantly suppressed Ang II-induced increases in the myocyte surface area, HW/BW, LVW/BW, SBP and left ventricular wall thickness. Furthermore, Pue significantly suppressed Ang II-induced increases in ANP, BNP and β-MHC expression in the left ventricles in vivo. Treatment of cardiomyocytes with Pue (50–500 μmol/L) did not affect the viability of cardiomyocytes in vitro. Pretreatment of cardiomyocytes with Pue dose-dependently inhibited Ang II-induced increases in ROS production, NF-κB binding activity, protein synthesis and cell breadth. Furthermore, pretreatment with Pue significantly suppressed Ang II-induced activation of ERK1/2, p38 and the NF-κB pathway proteins and the expression of ANP and β-MHC in cardiomyocytes. The positive drug valsartan exerted similar effects on Ang II-induced cardiac hypertrophy in vivo and in vitro.
Conclusion:
Pue attenuates Ang II-induced cardiac hypertrophy by inhibiting activation of the redox-sensitive ERK1/2, p38 and the NF-κB pathways.
Similar content being viewed by others

Introduction
Cardiac hypertrophy is an increase in the size and mass of the heart that ultimately leads to cardiac dysfunction and heart failure. This increase in size can be attributed to a number of diverse factors that include physiological, mechanical, hormonal, and genetic influences. An increasing number of studies suggest that the increased production of ROS is involved in the hypertrophic process1,2. ROS have been studied as a second messenger and recognized as an important trigger of both in vivo and in vitro hypertrophic responses to mechanical stretch or to hormonal stimuli, such as Ang II3. In last decade, increasing amounts of data have indicated that Ang II activates nicotinamide adenine dinucleotide phosphate (NADPH) oxidase via the Ang II receptor 1 (AT1), which leads to increased generation of ROS such as superoxide anion, hydrogen peroxide and the hydroxyl radical4,5. Moreover, ERK1/2, p38 MAPK and the NF-κB pathway are reported to be redox-sensitive and involved in cardiomyocyte hypertrophy6,7,8. Therefore, antioxidants may be a useful treatment modality for the inhibition of cardiac hypertrophy.
Pue (daidzein 8-C-glucoside, C21H20C9) is an isoflavone derived from Kudzu roots. Isoflavones are plant polyphenolic antioxidants that occur abundantly in soybean and the root of the Pueraria lobata plant. With its comprehensive pharmacological actions, Pue is clinically used in China for the treatment of cardiovascular diseases, including coronary artery disease, heart failure, hypertension and myocardial infraction9,10. A number of studies have demonstrated that Pue can block β-adrenoreceptors in isolated organs and the whole animal11, act as an anti-arrhythmic agent12, lower blood pressure and inhibit plasma renin activity in spontaneously hypertensive rats13 and protect ventricular myocytes against cardiac damage and arrhythmias by inhibiting recovery from the inactivation of cardiac Na+ channels14. Although Pue has been widely used in the treatment of cardiovascular diseases, its role in Ang II-induced cardiac hypertrophy remains unknown, and the effect of Pue on the generation of ROS and downstream signaling pathways in cardiomyocytes after Ang II stimulation has not been investigated. The aims of this study were therefore to determine whether Pue can attenuate Ang II-induced cardiac hypertrophy in vivo and in vitro and to identify the molecular mechanisms responsible for these putative effects.
Materials and methods
Animals and protocols
This animal study was approved by the Research Committee of Chinese Academy of Medical Science & Peking Union Medical College, and all animal experiments conform to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health. Eight-week-old, healthy, specific pathogen free grade, male C57BL/6J mice (n=40) weighing 19–22 g were purchased from Vital River Laboratories (SCXK jing 2011-0003, Beijing, China) and randomized into 4 groups: a sham group of vehicle-infused mice that received only PBS; an Ang II (Sigma-Aldrich, St Louis, CA, USA)-infused group; a Pue treatment group with ig administration of Pue (purity>99%, Shandong Fangming Pharmaceutical Co, product series: 1006301; Heze, China) at a dosage of 100 mg·kg−1·d−1 in water; a valsartan treatment group with ig administration of valsartan (Val, purity>99%, Novartis Pharmaceutical Co, product series: X1040; Beijing, China) at a dosage of 20 mg·kg−1·d−1 in water. All of the groups, except the sham group, simultaneously received Ang II (2.5 μg·kg−1·min−1, dissolved in PBS with 10 μmol/L acetic acid) from an implanted subcutaneous minipump (model 2002, Alza, Mountain View, CA, USA) for 15 d15. All mice had free access to drinking water and feed. This dose of Pue did not affect the growth or the food and water intake of the mice.
Morphological examination
After 15 d of treatment, mice were weighed and sacrificed. The thoracic cavity was immediately opened. With the blood squeezed out, the heart was excised and rinsed in cold physiological saline. The blood vessels and all the epicardial fat were removed. The heart was isolated, blotted, and weighed on an analytical balance. The HW/BW and LVW/BW ratios were calculated and used as an index of cardiac hypertrophy. Histopathology was performed by the Institute of Laboratory Animal Science, Chinese Academy of Medical Sciences & Peking Union Medical College. Immediately after gross dissection, the left ventricles were sectioned into 2–3 mm thick slices and immersed in a formalin solution for more than 7 d. The sections were then dehydrated in ethanol, cleared in dimethylbenzene and finally embedded in paraffin. The paraffin-embedded sections of the left ventricle were then cut into 5-μm-thick sections and stained with HE for light microscopic evaluation. A single myocyte was measured with the Image Pro-Plus, version 6.0, quantitative digital image analysis system (Media Cybernetics, Rockville, USA). The outline of 100 cardiomyocytes was traced in each group.
Echocardiography and systolic blood pressure assessment
After 15 d of treatment, echocardiography was performed using the Siemens Acuson CV-70 ultrasonographic system (Siemens Medical Solutions, Mountain View, CA, USA) as previously described16. Isoflurane (1.5% mixed with O2) was used to provide adequate sedation but minimal cardiac suppression during echocardiography. Mice were placed in a supine position, and a 13 MHz probe was applied to the left hemithorax. M-mode measurements of the left ventricular end-diastolic diameter, end-systolic diameter and wall thickness were performed according to the American Society of Echocardiography guidelines. The parameters obtained were as follows: LV end diastolic diameter (LVEDD), LV end systolic diameter (LVESD), LV posterior wall thickness (LVPWth) and interventricular septal wall thickness (IVSth). Fractional shortening (FS) was calculated as (LVEDD-LVESD)/LVEDD×100%.
After the implantation of the minipump, systolic blood pressure (SBP) was measured twice a week, using the tail-cuff method without anesthesia as previously described17. Mice were placed in restrainers, and their body temperature was maintained at 34 °C by a warming chamber. The IITC tail cuff sensor (IITC Life Science, Woodland Hills, CA, USA) containing both the inflation cuff and the photoelectric sensor was placed on the tail and attached to the restrainer. The cuff was inflated to a pressure of 200 mmHg and then deflated slowly. Upon reappearance of pulse signals, the IITC Life Science Blood Pressure System software would record, analyze and report the SBP data from the IITC amplifier.
Neonatal mouse primary cardiomyocyte culture
The procedure used to culture ventricular cardiomyocytes from neonatal mice was established by modifying previously described methods18. Primary cultures of cardiomyocytes were prepared from 1–2 d old neonatal C57BL/6J mice. Minced ventricular myocardium was placed into a pH 7.2 D-Hanks solution. The cells were dissociated by a solution of 0.125% trypsin without EDTA. After each of the several successive 5-min incubations, the dissociated cells were mixed with Dulbecco's modified Eagle's medium/F12 medium (DMEM/F12, Hyclone, Waltham, USA) containing 12.5% fetal bovine serum (FBS, Hyclone, Waltham, USA) and then centrifuged and pooled. The dissociated cells were enriched for cardiomyocytes by differential adhesion for 60 min and were plated at a density of 1.0×105 cells/cm2. Cultures were incubated in a humidified environment of 5% CO2–95% O2 at 37 °C. Bromodeoxyuridine (Sigma-Aldrich, St Louis, CA, USA) at 100 μmol/L was added into the medium to inhibit proliferation of non-myocytes. This procedure yielded cultures with 94%–98% myocytes, as assessed by cardiac specific anti-sarcomeric α-actinin (Sigma-Aldrich, St Louis, CA, USA) FITC immunohistochemistry. After 3 nights of incubation in DMEM/F12 containing 12.5% FBS, the attached cells were grown to 90% confluence and then rinsed and maintained in DMEM/F12 containing 0.1% FBS. After 24 h of serum starvation, the cardiomyocytes were pretreated for 2 h with Pue (50, 100, and 200 μmol/L), 10 μmol/L Val, 10 mmol/L N-acetylcysteine (NAC, Sigma-Aldrich, St Louis, CA, USA), 10 μmol/L apocynin (Apo, Sigma-Aldrich, St Louis, CA, USA), 10 μmol/L PD98059 (Sigma-Aldrich, St Louis, CA, USA), 10 μmol/L SB203580 (Sigma-Aldrich, St Louis, CA, USA) or 25 μmol/L pyrrolidine dithiocarbamate (PDTC, Sigma-Aldrich, St Louis, CA, USA) and were then stimulated with or without 1 μmol/L Ang II for the indicated times. The cells were then harvested and extracted for the analysis.
Cell immunohistochemistry
Cardiomyocytes were identified by immunostaining with an α-actinin antibody. Fused cells were fixed with 4% paraformaldehyde for 30 min at room temperature and then rinsed three times with 0.1 mol/L PBS, pH 7.4. Fixed cells were permeabilized with 0.1% Triton X-100 in PBS for 30 min, blocked with 1% BSA in PBS for 30 min and then rinsed three times with 0.1 mol/L PBS, pH 7.4. The cells were incubated with a primary antibody against α-actinin (diluted 1:500) over a 24-h period at 4 °C. After being washed with 0.1 mol/L PBS, they were then incubated with a FITC-conjugated IgG (Santa Cruz, Dallas, USA) (diluted 1:400) for 1 h at room temperature. The cells were then rinsed three times with PBS. After being rinsed, the specimens were mounted in PBS and observed with a fluorescence microscope (IX71-F22FL/PH, Olympus, Tokyo, Japan). The obtained cardiomyocyte cultures were greater than 96% purity.
Cell viability test and Pue evaluation of cytotoxicity
Cell viability was assessed using the MTT assay. The cells were seeded into a 96-well plate at a cell density of 8×103 cells/well for 24 h, followed by Pue treatment for 48 h. Next, 5 μL of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) at 5 mg/mL was added to the medium and incubated for 4 h at 37 °C. After removing the culture medium, 200 μL of dimethyl sulfoxide (DMSO) was added. The plates were read using an enzyme-linked immunosorbent assay plate reader (Bio-Rad, Hercules, USA) at 570 nm. The viability of the cells was assayed by the optical density value.
Measurement of in vivo and in vitro ROS production
The determination of intracellular ROS production was based on the oxidation of 2,7-dichlorofluorescin diacetate (H2DCFDA, Invitrogen, Carlsbad, USA) into a fluorescent 2,7-dichlorofluorescein (DCF). As previously described19 for the in vivo analysis, 300 mg of ventricle tissue was homogenized with a homogenizer for 20 s in 2 mL of a hypotonic lysis buffer [10 mmol/L KCl, 2 mmol/L MgCl2, 1 mmol/L dithiothreitol (DTT), 0.1 mmol/L EDTA, 0.1 mmol/L phenylmethylsulfonyl fluoride (PMSF), 1 μmol/L pepstatin, 2 μmol/L leupeptin, 20 mmol/L β-glycerophosphate, 20 mmol/L NaF and 2 mmol/L Na3VO4, 10 mmol/L hydroxyethyl piperazine ethane-sulfonic acid (HEPES), pH 7.4]. H2DCFDA at 25 μmol/L was added to the homogenates to bring the final volume to 250 μL. Changes in fluorescence intensity were measured every 5 min for 30 min on a GENios fluorescence plate reader (Tecan Instruments, Salzburg, Austria) with excitation and emission wavelengths set at 485 and 530 nm, respectively. For in vivo analysis, cells were loaded with a 5 μmol/L solution of H2DCFDA for 30 min at 37 °C in the dark. Cells were treated with Ang II in the presence or absence of Pue for indicated times. The cells were subsequently washed twice with PBS, detached by trypsin, harvested and then immediately analyzed with a BD FACSCalibur (BD Biosciences, San Diego, USA) cytometer with excitation and emission settings of 488 nm and 530 nm, respectively. For each sample, 10 000 cells were counted.
Cytosolic and nuclear extracts
Extracts were prepared as described by Finto et al20. The cells were washed in cold PBS, resuspended in 500 μL of cold buffer [10 mmol/L HEPES, 2 mmol/L MgCl2, 0.1 mmol/L EDTA, 10 mmol/L KCl, 1 mmol/L DTT, 0.5 mmol/L PMSF, 1 μg/mL leupepstatin, 1 μg/mL pepstatin and 1 μg/mL leucine thiol, 0.1% IgePal CA 630 pH 7.9] and left on ice for 30 min. The samples were then mixed and centrifuged at 4000×g for 30 min. The supernatant (cytosolic extract) was kept at −70 °C. The pellet was resuspended in 150 μL of a cold saline buffer [20 mmol/L HEPES, 50 mmol/L KCl, 0.1 mmol/L EDTA, 1.5 mmol/L MgCl2, 300 mmol/L NaCl, 25% (w/v) glycerol, 1 mmol/L DTT, 0.5 mmol/L PMSF, 1 μg/mL leupepstatin, 1 μg/mL pepstatin and 1 μg/mL leucine thiol, 0.2% IgePal CA630, pH 7.9] and left on ice for 1 h. After centrifuging at 12 000×g for 15 min at 4 °C, the supernatant (nuclear extract) containing the nuclear proteins was collected and stored at −70 °C. Protein content was measured using the Modified Lowry protein assay kit (Pierce, Rockford, USA).
Western blot analysis
Anti-phospho-specific p38, anti-phospho-specific ERK1/2, anti-phospho-specific IκBα, anti-ERK, and anti-p38 antibodies were purchased from Cell Signaling Technology (Danvers, USA). Anti-p65 NF-κB and anti-HistoneH1 antibodies were purchased from Santa Cruz Biotechnology (Dallas, USA). After various treatments, ventricular tissue or cardiomyocytes were lysed with lysis buffer containing 50 mmol/L Tris-HCl (pH 7.5), 250 mmol/L NaCl, 5 mmol/L EDTA, 50 mmol/L NaF, 1 mmol/L DTT, 1% TritonX-100, 1 mmol/L sodium orthovanadate and protease inhibitors. After centrifugation, the supernatant fraction was removed, and protein concentrations were determined using the Bio-Rad protein assay. Proteins were separated by SDS-PAGE and transferred onto a polyvinylidene fluoride membrane. After blocking with a 5% nonfat milk blocking buffer (PBS containing 0.1% Tween 20, pH 7.5), the membrane was incubated with the desired primary antibody overnight at 4 °C and then incubated with an appropriate peroxidase-conjugated secondary antibody. The density of immunoreactive bands was visualized using the ECL Plus Western Blotting Detection System (Piscataway, USA).
Semi-quantitative RT-PCR analysis
Total RNA was isolated from the ventricular tissue or cardiomyocytes using TRIzol according to the manufacturer's instructions (Invitrogen, Carlsbad, USA), and RT-PCR was then performed with the PrimeScriptTM RT reagent Kit (Takara, Dalian, China). The RNA samples were resuspended in DEPC H2O and quantified spectrophotometrically at 260 nm to be 100 ng/μL. All RNA isolates had an OD260/OD280 between 1.8 and 2.0, indicating clean RNA isolates. The RNA quality was also checked by 2.0% agarose gel electrophoresis with 1 μg/mL EB staining. Moloney murine leukemia virus reverse transcriptase was used with either oligo (dT) primers or random primers in a 20 μL reaction mixture to reverse transcribe 500 ng of the extracted RNA. The resulting cDNA was amplified by PCR for the ANP, BNP and β-MHC genes. The conditions used for the PCR were as follows: 94 °C 3 min; 94 °C 30 s; 50 °C 20 s; 72 °C 20 s; 72 °C 10 min; 32 cycles. The following primers were used for the PCR: atrial natriuretic peptide (ANP) forward primer: 5′-GTGTACAGTGCGGTGTCCAA-3′ and reverse primer: 5′-ACCTCATCTTCTACCGGCAT-3′; brain natriuretic peptide (BNP) forward primer: 5′-GAGGTCACTCCTATCCTCTGG-3′ and reverse primer: 5′-GCCATTTCCTCCGACTTTTCT-3′; β-myosin heavy chain (β-MHC) forward primer: 5′-GCATTCTCCTGCTGTTTCCTT-3′ and reverse primer: 5′-TGGATTCTCAAACGTGTCTAGTGA-3′ ; GAPDH forward primer: 5′-CGGAGTCAACGGATTTGGTCGTAT-3′ and reverse primer: 5′-AGCCTTCTCCATGGTGGTGAAGAC-3′ (Invitrogen, Carlsbad, USA).
EMSA
EMSAs were performed according to the manufacturer's instructions of Gel Shift Assay System Commercial Kit (Promega, Madison, USA). Double-stranded consensus oligonucleotide (5′-AGT TGA GGG GAC TTT CCC AGG C-3′) was 5′ end labeled with a T4 polynucleotide kinase and [γ-32P]ATP (Promega, Madison, USA). Ten micrograms of nuclear protein was pre-incubated for 10 min at room temperature in 9 μL of a binding buffer consisting of 10 mmol/L pH 7.5 Tris-HCl, 4% glycerol, 1 mmol/L MgCl2, 0.5 mmol/L EDTA, 0.5 mmol/L DTT, 0.5 mmol/L NaCl and 0.05 mg/mL poly(dI-dC). After the addition of the 32P-labled oligonucleotide probe, the incubation was continued for 20 min at room temperature. The reaction was stopped by adding 1 μL of a gel-loading buffer, and the protein-DNA binding complexes were analyzed with nondenaturing 4.8% polyacrylamide gel electrophoresis in 0.5×TBE buffer (44.5 mmol/L Tris, 44.5 mmol/L boric acid, and 1 mmol/L EDTA). After electrophoresis was conducted, the gel was vacuum dried and exposed to X-ray film (Fuji Hyperfilm, Tokyo, Japan) with an intensifying screen at −70 °C.
Cardiomyocyte surface area and volume assay
The cardiomyocyte surface area and volume were measured according to a previously described method21. Cell images were captured with a digital camera fixed to a microscope (IX71-F22FL/PH, Olympus, Tokyo, Japan). Cardiomyocyte surface area and volume were analyzed using Image Pro-Plus image analysis software and a CIAS-1000 image analyzing system (Daheng-Image, Beijing, China), respectively. One hundred cells from randomly selected fields in three wells were examined for each condition. Cardiomyocyte surface area and volume were determined after 48 h of stimulation with Ang II, and Pue-treated cells were compared with control cells.
Measurement of [3H]leucine incorporation
[3H]Leucine incorporation was measured as described previously22. Cells were grown in 24-well plates with serum-free medium for 24 h. Cells were then treated with Ang II for 24 h after pretreatment with or without Pue or other inhibitors. The cells were pulsed with 1 μCi/mL of [3H]leucine (Amersham Biosciences, Piscataway, USA) during the last 4 h before harvest. After being washed with PBS, cells were treated with 5% trichloroacetic acid for 30 min and then washed with PBS again. Finally, cells were solubilized in 500 μL of 1 mol/L NaOH. After neutralization with 0.5 mol/L HCl, an aliquot was taken to determine the level of incorporated radioactivity using the Beckman LS 3801 liquid scintillation counter (Beckman, Fullerton, USA).
Modified Lowry method for protein content assay
Cardiomyocytes were cultured in 12-well plates at a density of 5.0×105 cells/well for 24 h, and total protein content per well was measured by the modified Lowry protein assay kit (Pierce, Rockford, USA).
Statistical analysis
Data are expressed as mean±SEM. The significance in differences between groups was tested by either one-way analysis of variance (ANOVA) with the Tukey correction or unpaired t-tests for continuous variables using SPSS16.0 statistical software (SPSS, Chicago, USA). A two-tailed P<0.05 was considered statistically significant.
Results
Pue attenuates cardiac hypertrophy in vivo
After 15 d of experiment, Ang II-infused mice exhibited cardiac hypertrophy when compared with sham group, as evidenced by the classical increase in heart size and increases in the HW/BW and LVW/BW ratios. The percentage of animals with cardiac hypertrophy was 98.3%, as evidenced by classical increase in heart size, HW/BW and LVW/BW, when compared with sham group. In the Pue-treated mice, the Ang II-induced increases in heart size and the HW/BW and LVW/BW ratios were markedly decreased to levels similar to that of sham or Val-treated mice. Under the microscope, the left ventricular tissues from Ang II-infused mice exhibited obvious cardiomyocyte hypertrophy, and the cardiomyocyte surface area was significantly larger than that of the sham group. These characteristic changes induced by Ang II were markedly attenuated by long-term treatment with Pue at 100 mg/kg (Figure 1A and 1B). Treatment with Pue or Val significantly attenuated the observed increase in expression levels in the Ang II-infused groups (Figure 1C). Treatment with Pue or Val prevented both the decreases in LVESD and LVEDD and the increases in IVSth, LVPWth and FS that were observed in the Ang II-infused group that did not receive any treatment (Figure 2A). The SBP was significantly higher in the Ang II-infused mice than sham mice during Ang II-perfusion, and this increase in SBP was markedly decreased with Pue or Val treatment (Figure 2B).

The in vivo inhibitory effect of Pue on cardiac hypertrophy. (A) Representative cross-sections of the left ventricles stained with HE (original magnification×200) and gross specimens of the hearts. (B) Bar graph shows the quantitative analysis of HW/BW, LVW/BW, and mean cell area. (C) The expression of fetal-type genes in the left ventricles randomly picked from each group. Mean±SEM. n=3. cP<0.01 vs sham group. fP<0.01 vs Ang II-infused group.

Noninvasive assessment of cardiac hypertrophy and SBP and measurements of ROS production in vivo. (A) Representative M-mode images of cardiac hypertrophy assessed by echocardiography after 15 d of Ang II infusion. (B) Change in the SBP during Ang II-perfusion in mice. (C) Measurements of ROS production induced by Ang II in the left ventricle homogenates. Mean±SEM. n=3. cP<0.01 vs sham group. fP<0.01 vs Ang II-infused group.
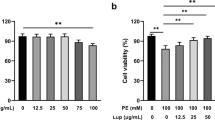
Effects of Pue on cardiomyocyte viability.
Exposure of cultured cardiomyocytes to Pue for 48 h did not have any significant effect on cardiomyocyte viability. Additionally, myocyte monolayers continued to contract synchronously in the presence of the different doses of Pue (Figure 3).

Effects of Pue on the viability of cardiomyocytes. Mean±SEM. n=3.
In vitro measurements of ROS and signaling proteins induced by Ang II
Ang II increased the intracellular ROS production, the phosphorylated levels of ERK1/2, p38, and IκBα proteins and the translocation of p65 NF-κB into the nucleus in a time-dependent manner (Figure 4). The optimal response was achieved at 4 h, and the cells were therefore stimulated with Ang II for 4 h in subsequent experiments.

Measurements of ROS production and the activation of signaling proteins induced by Ang II in vitro. (A) The time course of ROS generation. (B) The levels of phosphorylated ERK1/2, p38, and IκBα and p65 NF-κB translocation into the nucleus over time (Immunoblot analysis of p65 NF-κB in cytosolic (C) and nuclear (N) extracts from cells). Mean±SEM. n=3. cP<0.01 vs control.
The in vivo and in vitro effects of Pue on the intracellular excess production of ROS
In vivo infusion of Ang II triggered an increase in ROS levels, which was decreased by Pue (Figure 2C). In vitro ROS generation was also inhibited by Pue in a dose-dependent manner similar to the inhibitory effects observed with NAC, Val, or Apo. Additionally, we treated cells with 100 μmol/L exogenous H2O2 for 1 h as a positive control. Our results showed that both Pue and NAC can scavenge intracellular H2O2 (Figure 5).

Inhibitory effects of Pue on the Ang II or exogenous H2O2-induced intracellular ROS generation. Mean±SEM. n=3. cP<0.01 vs control group. fP<0.01 vs Ang II-treated group. iP<0.01 vs H2O2-treated group.
Blockade of ERK1/2, p38, and NF-κB pathway activation by Pue
To further explore the molecular mechanisms through which Pue impairs the cardiac hypertrophic response, we examined the in vitro effects of Pue on the ERK1/2, p38, and NF-κB pathways. Ang II induced a marked increase in the phosphorylated levels of ERK1/2, p38, and IκBα. However, Pue treatment blocked the activation of ERK1/2, p38, and IκBα in a dose-dependent manner. Similarly, the activation of these signaling proteins mediated by Ang II treatment was also attenuated by NAC and Apo (Figure 6A). In line with the in vitro findings, we found that marked in vivo phosphorylation of ERK1/2, p38, and IκBα was almost completely blocked by Pue (Figure 6B). To determine whether these pathways were redox sensitive, we exposed the cells to either Ang II or 100 μmol/L exogenous H2O2 to examine the effect of NAC on individual ERK1/2, p38, and IκBα phosphorylation. H2O2 treatment for 1 h directly activated ERK1/2, p38, and IκBα, and this activation was suppressed by NAC and Pue (Figure 6C). Moreover, Ang II-induced NF-κB translocation into the nucleus was prevented by Pue, NAC, and Apo (Figure 7B). An EMSA was also performed to investigate whether Pue interferes with NF-κB binding activity. The results of these shift assays confirmed the presence of p65 NF-κB in the protein complex and demonstrated that Pue could inhibit this binding activity in a dose-dependent manner. In addition, we measured Ang II-induced IκBα degradation in a time-dependent manner. After a 2 h pretreatment with either PD98059 or SB203580, the cells were stimulated by Ang II for 4 h. IκBα degradation was markedly inhibited by both PD98059 and SB203580, suggesting that IκBα degradation was partially MAPK dependent (Figure 7A). The results therefore indicated that blocking the p38 or ERK1/2 signaling pathways with SB203580 or PD98059 attenuated the Ang II-induced increase in NF-κB nuclear translocation and DNA binding activity (Figure 7B, 7C).

Inhibitory effects of Pue on the Ang II or exogenous H2O2-induced ERK1/2, p38, and IκBα activation. (A) The effects of Pue, NAC, and Apo on the Ang II-induced ERK1/2, p38, IκBα activation in vitro. (B) The effects of Pue on the ERK1/2, p38, and IκBα activation in the left ventricle tissues (n=3) randomly picked from each group. (C) The effects of Pue and NAC on the H2O2-induced ERK1/2, p38, and IκBα activation. Mean±SEM. n=3. cP<0.01 vs control or sham group. fP<0.01 vs Ang II-treated group. iP<0.01 vs H2O2-treated group.

Inhibitory effects of Pue on Ang II-induced NF-κB translocation and DNA binding activity. (A) The time-dependent manner of IκBα protein degradation and the effects of PD98059 and SB203580 on IκBα degradation. (B) The effects of Pue, NAC, Apo, PD98059, and SB203580 on Ang II-induced p65 NF-κB nuclear translocation. (C) The effects of Pue, NAC, PD98059, and SB203580 on the DNA binding activity of NF-κB. Mean±SEM. n=3. bP<0.05, cP<0.01 vs control group. eP<0.05, fP<0.01 vs Ang II-treated group.
Effects of Pue on the Ang II-induced cultured cardiomyocyte hypertrophy
Cardiomyocyte hypertrophy is characterized by an increase in cell size, protein synthesis and the expression of fetal-type genes. We therefore examined the effects of Pue on these parameters. Cardiomyocytes were pretreated with Pue for 2 h before stimulation with Ang II. The hypertrophic response was determined after 48 h. Ang II induced a marked increase in either cell surface area or volume, which was partially inhibited by Pue (Figure 8A). Moreover, the increase in [3H]leucine incorporation and total protein content detected after 24 h of culture in the presence of Ang II was prevented in cells preincubated with Pue (Figure 8C). Further, Pue suppressed the Ang II-induced expression of the ANP and β-MHC genes for 24 h (Figure 8B). In addition, we used NAC, SB203580, PDTC, and PD98059 to determine whether the inhibition of Ang II-induced cardiomyocyte hypertrophy by Pue is via the attenuation of ROS and the activation of the downstream ERK1/2, p38 or NF-κB pathways. The results showed that NAC, SB203580, PDTC, and PD98059 could markedly reduce the Ang II-induced protein synthesis and fetal-type gene mRNA expression in cardiomyocytes (Figure 8B, 8C).

Effect of Pue on the Ang II-induced hypertrophic response in cultured cardiomyocytes. (A) Representative images of cultured cardiomyocytes (original magnification ×400). The bar graph shows the quantified cardiomyocyte surface area and volume. (B) Expression of fetal-type genes in the cardiomyocytes. (C) The rate of protein synthesis and the total protein content in the cardiomyocytes. Mean±SEM. n=3. cP<0.01 vs control group. fP<0.01 vs Ang II-treated group.
Discussion
The major outcome of this study clearly shows for the first time that Pue inhibits Ang II-induced cardiac hypertrophy in vivo and in vitro. These findings are supported by the following observations. In animal experiments, we used a minipump to infuse Ang II into the mice to establish cardiac hypertrophy. The characteristic hypertrophic changes, such as increases in the HW/BW and LVW/BW ratios, the enlargement of cardiomyocytes, the reappearance of fetal isoforms of cardiac genes and increases in the chamber size and wall thickness, were markedly inhibited after a long treatment with 100 mg·kg−1·d−1 Pue. In line with the in vivo findings, we found in cultured cardiomyocytes that Pue also significantly depressed the Ang II-induced marked increase in the cell size and inhibited the induction of fetal-type gene expression and protein synthesis. Then, we further explored in cultured cardiomyocytes the molecular mechanisms through which Pue inhibits cardiac hypertrophy. Although the mechanism by which Ang II induces cardiomyocyte hypertrophy is not fully understood, members of the MAPK family, PI3Kα-Akt, Jak-STAT, the Na+/H+ exchanger, AMP-activated protein kinase (AMPK) and DNA-binding transcription factors, such as NF-κB and AP-1, all reportedly play a pivotal role in the development of cardiac hypertrophy23,24. Among them, ERK1/225, p3826, and NF-κB27,28 have been noted as the essential regulators of the hypertrophic response. With this mind, the inhibitory mechanisms of Pue on cardiac hypertrophy were examined by investigating its effect on ERK1/2, p38, and NF-κB activation. We observed that Ang II enhanced ERK1/2, p38, and IκBα activation in a time-dependent manner, which was almost completely blocked by Pue, NAC, and Apo. The inhibitory effect of Pue on these signaling proteins may be via its ability to act as an antioxidant because mounting evidence has suggested that ROS play an important role in Ang II-induced cardiac hypertrophy and that increased ROS generation leads to the activation of downstream signaling pathways and transcription factors, including ERK1/2, p38, and NF-κB29,30,31. Our present study showed that following Ang II stimulation, intracellular production of ROS increased in a time-dependent manner, and this effect was abolished not only by Pue in a dose-dependent manner but also by Val and Apo treatment, which indicates that Ang II activates NAD(P)H oxidase to generate intracellular ROS via an AT1 receptor. In an additional Western blot analysis, we found that the upregulation of phosphorylated ERK1/2, p38, and IκBα levels by Ang II was significantly downregulated by Apo and NAC. Moreover, these proteins were also directly activated by exogenous H2O2, suggesting that these signaling pathways are redox-sensitive, which could be a potential site of action for Pue in cardiomyocytes. Pue acts as an antioxidant through multiple mechanisms, such as scavenging free radicals, increasing superoxide dismutase and catalase activity and decreasing malondialdehyde32,33,34. Our results also demonstrated its potential to scavenge intracellular H2O2. Being concordant to previous findings indicating that many antioxidants can inhibit the activation of the MAPK and NF-κB pathways by scavenging ROS35,36,37, it is therefore not surprising that Pue interfered with Ang II-mediated ROS generation and also blocked associated downstream signaling pathways. Finally, Pue inhibited NF-κB activation by disrupting DNA-binding and transcriptional activity in the nucleus. It partly did this by downregulating the levels of phosphorylated IκBα and then inhibiting p65 NF-κB translocation into the nucleus. Increasing evidence shows that some members of the MAPK family participate in the activation of the NF-κB pathway38,39. Therefore, we examined the role of ERK1/2 and p38 in Ang II-induced activation of NF-κB binding. It is of interest that SB203580 and PD98059 as specific inhibitors of p38 and ERK1/2 activation, respectively, partially inhibited IκBα degradation and then disrupted Ang II-induced NF-κB nuclear translocation, DNA-binding and transcriptional activity, suggesting that NF-κB activation is at least partly dependent on the ERK1/2 and p38 signaling pathways40,41,42. To further elucidate the significance of the ERK1/2, p38, and NF-κB pathways in the anti-hypertrophic effect of Pue, we found that the Ang II-induced increase in [3H]leucine incorporation, cell size and ANP and β-MHC gene expression was significantly decreased by SB203580, PD98059, and PDTC. This finding confirms that the ERK1/2, p38, and NF-κB pathways play significant roles in the hypertrophic effect of Ang II.
Taken together, the present study delivers important new insights into the molecular mechanisms of action of Pue on cardiomyocyte hypertrophy. These data suggest that Pue suppresses cardiac hypertrophy by blocking the excessive generation of ROS and disrupting the downstream activation of the ERK1/2, p38, and NF-κB pathways.
Author contribution
Gang CHEN and Qi-yang HE designed the research; Gang CHEN, Cong SHEN, Xiu-ming ZHANG, and Shi-qi PAN performed the research; Gang CHEN and Shi-qi PAN contributed new analytical tools and reagents; Gang CHEN and Cong SHEN analyzed the data; Gang CHEN, Shi-fen PAN, and Qi-yang HE wrote and revised the paper.
References
Bendall JK, Cave AC, Heymes C, Gall N, Shah AM . Pivotal role of a gp91 (phox)-containing NAD(P)H oxidase in angiotensinII-induced cardiac hypertrophy in mice. Circulation 2002; 105: 293–6.
Cai H, Griendling KK, Harrison DG . The vascular NAD(P)H oxidases as therapeutic targets in cardiovascular diseases. Trends Pharmacol Sci 2003; 24: 471–8.
Frank GD, Eguchi S, Yamakawa T, Tanaka S, Inagami T, Motley ED . Involvement of reactive oxygen species in the activation of tyrosine kinase and extracellular signal-regulated kinase by angiotensinII. Endocrinology 2000; 141: 3120–6.
Garrido AM, Griendling KK . NADPH oxidases and angiotensinII receptor signaling. Mol Cell Endocrinol 2009; 302: 148–58.
Li YQ, Li XB, Guo SJ, Chu SL, Gao PJ, Zhu DL, et al. Apocynin attenuates oxidative stress and cardiac fibrosis in angiotensinII-induced cardiac diastolic dysfunction in mice. Acta Pharmacol Sin 2013; 34: 352–9.
Koichi T, Masaaki H, Toshikazu T . Redox regulation of MAPK pathways and cardiac hypertrophy in adult rat cardiac myocyte. J Am Coll Cardiol 2001; 37: 676–85.
Freund C, Schmidt-Ullrich R, Baurand A, Dunger S, Schneider W, Loser P, et al. Requirement of nuclear factor-kappaB in angiotensinII-and isoproterenol-induced cardiac hypertrophy in vivo. Circulation 2005; 111: 2319–25.
Higuchi Y, Otsu K, Nishida K, Hirotani S, Nakayama H, Yamaguchi O, et al. Involvement of reactive oxygen species-mediated NF-kappa B activation in TNF-alpha induced cardiomyocyte hypertrophy. J Mol Cell Cardiol 2002; 34: 233–240.
Yue HW, Hu XQ . Pharmacologic value of radix Puerariae and puerarine on cardiovascular system. Zhongguo Zhong Xi Yi Jie He Za Zhi 1996; 16: 382–84. Chinese.
Zhang Z, Lam TN, Zuo Z . Radix puerariae: an overview of its chemistry, pharmacology, pharmacokinetics, and clinical use. J Clin Pharmacol 2013; 53: 787–11.
Lu XR, Gao E, Xu LZ, Li HZ, Kang B, Chen WN, et al. Blocking effect of puerarin on beta-adrenoceptors of isolated organs and the whole animal. Acta Pharmacol Sin 1986; 7: 537–9.
Chai XS, Wang ZX, Chen PP, Wang LY, Lv XR, Kang B . Anti-arrhythmic action of puerarin. Acta Pharmacol Sin 1985; 6: 166–8.
Song XP, Chen PP, Chai XS . Effects of puerarin on blood pressure and plasma renin activity in spontaneously hypertensive rats. Acta Pharmacol Sin 1988; 9: 55–8.
Zhang GQ, Hao XM, Dai DZ, Fu Y, Zhou PA, Wu CH . Puerarin blocks Na+ current in rat ventricular myocytes. Acta Pharmacol Sin 2003; 24: 1212–6.
Dai WJ, Chen HY, Jiang JD, Kong WJ, Wang YG . Silencing MR-1 attenuates inflammatory damage in mice heart induced by Ang II. Biochem Biophys Res Commun 2010; 391: 1573–8.
Shiomi T, Tsutsui H, Ikeuchi M, Matsusaka H, Hayashidani S, Suematsu N, et al. Streptozotocin-induced hyperglycemia exacerbates left ventricular remodeling and failure after experimental myocardial infarction. J Am Coll Cardiol 2003; 42: 165–72.
Whitesall SE, Hoff JB, Vollmer AP, D'Alecy LG . Comparison of simultaneous measurement of mouse systolic arterial blood pressure by radiotelemetry and tail-cuff methods. Am J Physiol 2004; 286: H2408–15.
Fei AH, Cao Q, Chen SY, Wang HR, Wang FL, Pan SM, et al. Salvianolate inhibits reactive oxygen species production in H2O2-treated mouse cardiomyocytes in vitro via the TGFβ pathway. Acta Pharmacol Sin 2013; 34: 496–500.
Kim JM, Heo HS, Ha YM, Ye BH, Lee EK, Choi YJ, et al. Mechanism of Ang II involvement in activation of NF-κB through phosphorylation of p65 during aging. AGE 2012; 34: 11–25.
Finco TS, Beg AA, Baldwin AS Jr . Inducible phosphorylation of IκBα is not sufficient for its dissociation from NF-κB and is inhibited by protease inhibitors. Proc Natl Acad Sci U S A 1994; 91: 11884–8.
Lu QM, Liu ZY . Effects of glucose flunctuation on cardiomyocyte hypertrophy in neonatal rats. Chin J Histochem Cytochem 2011; 20: 334–8.
Nakamura K, Fushimi K, Kouchi H, Mihara K, Miyazaki M, Ohe T . Inhibitory effects of antioxidants on neonatal rat cardiac myocyte hypertrophy induced by tumor necrosis factor alpha and angiotensin II. Circulation 1998; 98: 794–9.
Nozato T, Ito H, Tamamori M, Adachi S, Abe S, Marumo F, et al. G1 cyclins are involved in the mechanism of cardiac myocyte hypertrophy induced by angiotensin II. Jpn Circ J 2000; 64: 595–601.
Das S, Otani H, Maulik N, Das DK . Redox regulation of angiotensinII preconditioning of the myocardium requires MAP kinase signaling. J Mol Cell Cardiol 2006; 41: 248–55.
Hirotani S, Otsu K, Nishida K, Higuchi Y, Morita T, Nakayama H, et al. Involvement of nuclear factor-kappaB and apoptosis signal-regulating kinase1 in G-protein-coupled receptor agonist-induced cardiomyocyte hypertrophy. Circulation 2002; 105: 509–15.
Bueno OF, De Windt LJ, Tymitz KM, Witt SA, Kimball TR, Klevitsky R, et al. The MEK1-ERK1/2 signaling pathway promotes compensated cardiac hypertrophy in transgenic mice. EMBO J 2000; 19: 6341–50.
Liao P, Georgakopoulos D, Kovacs A, Zheng M, Lerner D, Pu H, et al. The in vivo role of p38 MAP kinases in cardiac remodeling and restrictive cardiomyopathy. Proc Natl Acad Sci U S A 2001; 98: 12283–8.
Purcell NH, Tang G, Yu C, Mercurio F, DiDonato JA, Lin A . Activation of NF-kappa B is required for hypertrophic growth of primary rat neonatal ventricular cardiomyocytes. Proc Natl Acad Sci U S A 2001; 98: 6668–73.
Kawano S, Kubota T, Monden Y, Kawamura N, Tsutsui H, Takeshita A, et al. Blockade of NF-κB ameliorates myocardial hypertrophy in response to chronic infusion of angiotensin II. Cardiovasc Res 2005; 67: 689–98.
Shih NL, Cheng TH, Loh SH, Cheng PY, Wang DL, Chen YS, et al. Reactive oxygen species modulate angiotensin II-induced beta-myosin heavy chain gene expression via Ras/Raf/extracellular signal-regulated kinase pathway in neonatal rat cardiomyocytes. Biochem Biophys Res Commun 2001; 283: 143–48.
Sano M, Fukuda K, Sato T, Kawaguchi H, Suematsu M, Matsuda S, et al. ERK and p38 MAPK, but not NF-kappaB, are critically involved in reactive oxygen species-mediated induction of IL-6 by angiotensin II in cardiac fibroblasts. Circ Res 2001; 89: 661–9.
Meng Z, Yan C, Deng Q, Gao DF, Niu XL . Curcumin inhibits LPS-induced inflammation in rat vascular smooth muscle cells in vitro via ROS-relative TLR4-MAPK/NF-κB pathways. Acta Pharmacol Sin 2013; 34: 901–11.
Chung MJ, Sung NJ, Park CS, Kweon DK, Mantovani A, Moon TW, et al. Antioxidative and hypocholesterolemic activities of water-soluble puerarin glycosides in HepG2 cells and in C57 Bcl/6J mice. Eur J Pharmacol 2008; 578: 159–70.
Liu CM, Ma JQ, Sun YZ . Puerarin protects the rat liver against oxidative stress-mediated DNA damage and apoptosis induced by lead. Exp Toxicol Pathol 2012; 64: 575–82.
Xiong FL, Sun XH, Gan L, Yang XL, Xu HB . Puerarin protects rat pancreatic islets from damage by hydrogen peroxide. Eur J Pharmacol 2006; 529: 1–7.
Li YQ, Zhang ZX, Xu YJ, Ni W, Chen SX, Yang Z, et al. N-Acetyl-L-cysteine and pyrrolidine dithiocarbamate inhibited nuclear factor-kappaB activation in alveolar macrophages by different mechanisms. Acta Pharmacol Sin 2006; 27: 339–46.
Nakamura K, Fushimi K, Kouchi H, Mihara K, Miyazaki M, Ohe T . Inhibitory effects of antioxidants on neonatal rat cardiac myocyte hypertrophy induced by tumor necrosis factor-alpha and angiotensinII. Circulation 1998; 98: 794–9.
Zou XJ, Yang L, Yao SL . Propofol depresses angiotensinII-induced cardiomyocyte hypertrophy in vitro. Exp Biol Med 2008; 233: 200–8.
Shen DF, Tang QZ, Yan L, Zhang Y, Zhu LH, Wang L, et al. Tetrandrine blocks cardiac hypertrophy by disrupting reactive oxygen species-dependent ERK1/2 signalling. Br J Pharmacol 2010; 159: 970–81.
Wang JX, Hou LF, Yang Y, Tang W, Li Y, Zuo JP . SM905, an artemisinin derivative, inhibited NO and pro-inflammatory cytokine production by suppressing MAPK and NF-kappaB pathways in RAW 264.7 macrophages. Acta Pharmacol Sin 2009; 30: 1428–35.
Yang XS, Liu SA, Liu JW, Yan Q . Fucosyltransferase IV enhances expression of MMP-12 stimulated by EGF via the ERK1/2, p38, and NF-κB pathways in A431 cells. Asian Pac J Cancer Prev 2012; 13: 1657–62.
Sée V, Rajala NK, Spiller DG, White MR . Calcium-dependent regulation of the cell cycle via a novel MAPK — NF-κB pathway in Swiss 3T3 cells. J Cell Biol 2004; 166: 661–72.
Acknowledgements
The work was supported by grants from the National S&T Major Special Project on Major New Drug Innovation (2012ZX09301002-001-015) and the National Natural Science Foundation of China (81273553).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Chen, G., Pan, Sq., Shen, C. et al. Puerarin inhibits angiotensin II-induced cardiac hypertrophy via the redox-sensitive ERK1/2, p38 and NF-κB pathways. Acta Pharmacol Sin 35, 463–475 (2014). https://doi.org/10.1038/aps.2013.185
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2013.185
Keywords
This article is cited by
-
Ameliorated biomechanical properties of carotid arteries by puerarin in spontaneously hypertensive rats
BMC Complementary Medicine and Therapies (2021)
-
Puerarin ameliorated pressure overload-induced cardiac hypertrophy in ovariectomized rats through activation of the PPARα/PGC-1 pathway
Acta Pharmacologica Sinica (2021)
-
Angiotensin II receptor blocker LCZ696 attenuates cardiac remodeling through the inhibition of the ERK signaling pathway in mice with pregnancy-associated cardiomyopathy
Cell & Bioscience (2019)
