
Abstract
Aim:
To investigate the effects of a new derivative of bisphosphonates, [2-(6-aminopurine-9-yl)-1-hydroxy-phosphine acyl ethyl] phosphonic acid (CP), on human gastric cancer.
Methods:
Human gastric cancer cell lines (SGC-7901, BGC-823, MKN-45, and MKN-28) and human colon carcinoma cell lines (LoVo and HT-29) were tested. Cell growth was determined using the MTT assay. Flow cytometry, Western blot, caspase activity assay and siRNA transfection were used to examine the mechanisms of anticancer action. Female BALB/c nude mice were implanted with SGC-7901 cells. From d6 after inoculation, the animals were injected with CP (200 μg/kg, ip) or vehicle daily for 24 d.
Results:
CP suppressed the growth of the 6 human cancer cell lines with similar IC50 values (3239 μmol/L). In SGC-7901 cells, CP arrested cell cycle progression at the G2/M phase. The compound activated caspase-9, increased the expression of pro-apoptotic proteins Bax and Bad, decreased the expression of anti-apoptotic protein Bcl-2. Furthermore, the compound selectively activated ERK1/2 without affecting JNK and p38 in SGC-7901 cells. Treatment of SGC-7901 cells with the specific ERK1/2 inhibitor PD98059 or ERK1/2 siRNA hampered CP-mediated apoptosis. In the human gastric cancer xenograft nude mouse model, chronic administration of CP significantly retarded the tumor growth.
Conclusion:
CP is a broad-spectrum inhibitor of human carcinoma cells in vitro, and it also exerts significant inhibition on gastric cancer cell growth in vivo. CP induces human gastric cancer apoptosis via activation of the ERK1/2 signaling pathway.
Similar content being viewed by others

Introduction
Surgery and chemotherapy are the most effective therapies for advanced gastric cancer. Although current chemotherapy programs continue to improve, the efficacy of presently available drugs is limited, and new treatments are urgently needed, particularly anticancer compounds that can target multiple steps in the apoptotic cascade to achieve auxiliary effects. Bisphosphonates (BPs) have been shown to reduce breast and prostate cancer cell invasion1,2 and to confer anti-angiogenesis activity3. Furthermore, BPs appear to have direct pro-apoptotic effects on cancer cells4; however, different breast cancer cell lines have varying sensitivities5,6. A recent clinical study demonstrated that adjuvant therapy with the amino-BP zoledronic acid could improve the disease-free survival of women with early breast cancer without bone metastases7. The anti-tumor activity of ibandronate has been illustrated preclinically in many types of cancer, including breast8, lung9, multiple myeloma10 and osteosarcoma11. Overall, the positive effects of BPs and their clinical potential in cancer have been widely confirmed both in vitro and in vivo12,13.
In a previous study, zoledronic acid was demonstrated to cause dose- and time-dependent inhibition of gastric cancer cell proliferation by inducing cellular apoptosis. The mechanism of this apoptosis induction was presumed to occur through the inhibition of Ras protein prenylation14. Zoledronic acid, which has been shown to have direct anti-proliferative and apoptotic effects, has also exhibited synergistic effects with gemcitabine and oxaliplatin in chemotherapy of metastatic gastric cancer15. However, to date, no clear molecular mechanism has been proposed to explain cellular changes in gastric cancer cells in response to BP treatment.
In the current study, we synthesized the compound [2-(6-amino-purine-9-yl)-1-hydroxy-phosphine acyl ethyl] phosphonic acid (CP), which showed obvious anti-proliferative activity against several human cancer cell types in preliminary studies. The ability of CP to inhibit the viability of gastric cancer cells and its underlying mechanism of action were examined in vitro. CP was found to induce apoptosis of human gastric cancer cells. The therapeutic potential of CP was also investigated using a xenograft nude mouse model. The study enhances our understanding of the molecular mechanism of CP-induced apoptosis and will aid the development of potential therapeutic agents for gastric cancer.
Materials and methods
Reagents
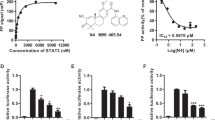
CP was provided by Ye JIANG from the School of Pharmaceutical Chemistry, Hebei Medical University. Detailed information on CP can be found at the State Intellectual Property Office of the People's Republic of China (patent number 200710185250.7)16. CP was originally synthesized to improve the efficacy and pharmacological characteristics of BP by substitution at a side chain (Figure 1A). Purified CP was dissolved in NaHCO3 at a concentration of 1000× and added to cells in exponential growth.

CP inhibits the growth of various gastric cancer cells. (A) Chemical structure of CP. (B) Effects of CP on the viability of various cancer cell lines. After treatment with CP for 24 h, cell viability was determined by the MTT assay, and the IC50 was calculated (n=6). (C) The cells were treated with 10, 20, 40, or 80 μmol/L CP, and cell growth was determined using the MTT assay at 1, 3, 6, 12, and 24 h (n=6). The results are presented as the mean±SEM from three independent experiments.
Small interfering RNAs (siRNAs) specific for human extracellular signal-regulated kinase1/2 (ERK1/2) mRNA and control siRNA were obtained from Cell Signaling Technology (Beverly, MA, USA). Antibodies specific for poly (ADP-ribose) polymerase (PARP), raf-1, phospho-raf-1, MEK1/2, phospho-MEK1/2, c-Jun NH2-terminal kinase (JNK), phospho-JNK, ERK1/2, phospho-ERK1/2, p38 mitogen-activated protein (MAP) kinase (p38) and phospho-p38 were also obtained from Cell Signaling Technology (Beverly, MA, USA). Antibodies against caspase-3, caspase-9, Bcl-2, Bax, and Bad as well as secondary antibodies were obtained from Abcam Biotechnology (Abcam, Cambridge, UK). Lipofectamine 2000, RPMI-1640, penicillin and streptomycin were purchased from Invitrogen (Carlsbad, CA, USA).
Cell lines
The human gastric cancer cell lines (SGC-7901, BGC-823, MKN-45, and MKN-28) and human colon carcinoma cell lines (LoVo and HT-29) were purchased from the American Type Culture Collection (Manassas, VA, USA) and maintained in RPMI-1640 supplemented with 10% fetal bovine serum (v/v). Normal human gastric epithelial cells (GES-1) were cultured and characterized as described previously17. The cells were incubated at 37 °C with 5% CO2 and subcultured every 4 d. The experiments were performed when the cells reached 60% to 80% confluency.
MTT assays
The effect of CP on the proliferation of cancer cells was measured using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Cells were cultured in 96-well plates (approximately 1×104) and incubated in growth medium for 24 h. The cells were then treated with various concentrations of CP for 1, 3, 6, 12, and 24 h. After drug treatment, the cells were incubated for 4 h in a medium containing 10 μL of MTT solution [dissolved in phosphate-buffered saline(PBS) at 5 mg/mL and sterilized]. The reaction was terminated by adding 100 μL of dimethyl sulfoxide (DMSO) to the medium and incubating for 10 min at 37 °C. The absorbance was measured at 570 nm in enzyme-linked immunosorbent assay (ELISA) reader to assess the cell viability. The experiments were repeated at least three times, and the data are expressed as the means±SEM.
Animal model
SGC-7901 cells were trypsinized and collected by centrifugation. Cell viability was confirmed to be above 95% based on trypan blue staining. The cells (2×106) were suspended in 0.2 mL of PBS and injected subcutaneously into the left armpit region of female BALB/c nude mice (age, 4 weeks; body weight, 15–17 g; grade, specific pathogen free [SPF]/viral antibody free [VAF]; certificate number, scsk (Jing) 2012-0001). After inoculation, the mice were kept in a limited access area at a controlled room temperature with food and water provided ad libitum. The mice were divided into two groups for different experiments (vehicle only and 200 μg/kg body weight CP). The mice were sacrificed after 30 d of treatment when the control tumors reached approximately 1400 mm3. The study was performed in accordance with the Guidelines of Animal Experiments from the Committee of Medical Ethics, National Health Department of China.
Cell cycle analysis
Cells were plated in 100-mm dishes at a density of 3×106 per dish. The medium was removed and pooled with trypsinized adherent cells. After the cells were washed and resuspended in cold PBS at a density of (1−3)×106 cells/mL, propidium iodide (PI) buffer (500 μL of 0.1 mg/mL PI in PBS) and RNase A (500 μL of a 2 mg/mL solution) were added. After 30 min of incubation at room temperature in the dark, the cells were filtered through a nylon mesh filter and analyzed by flow cytometry.
Annexin V-FITC/PI double-labeled flow cytometry
For detection of apoptotic cells treated with CP, the expression of annexin V-FITC and exclusion of PI were detected by two-color flow cytometry. After treatment with 40 μmol/L CP for the indicated times, SGC-7901 cells were washed and resuspended in PBS. Apoptotic cells were identified using the Annexin V-FITC Apoptosis Detection Kit (BD Biosciences, San Diego, CA, USA) according to the manufacturer's instructions. Early apoptotic cells were labeled as annexin V-positive and PI-negative by flow cytometric analysis.
ELISA
Cellular apoptosis was quantified by DNA fragmentation using the Cell Death Detection ELISAPLUS Kit (Roche Diagnostics, Mannheim, Germany). Briefly, SGC-7901 cells seeded in 96-well plates were treated with 40 μmol/L CP. After 0, 6, 12, or 24 h, the cells were lysed in 200 μL of lysis buffer, and 20 μL of the supernatant was reacted with 80 μL of anti-DNA immunocomplex conjugated with peroxidase, which binds to nucleosomal DNA, and anti-histone-biotin, which interacts with streptavidin-coated wells, in a microtiter plate for 2 h. At the end of the incubation, 100 μL of substrate was added, and color development was quantified at a wavelength of 405 nm. The results were calculated as the ratio of the absorbance of the CP-treated cells to the absorbance of the vehicle-treated cells.
Colorimetric assay for caspase activity
After the cells were treated with CP, the activities of caspase-3 and -9 were determined using a caspase colorimetric assay kit (Promega, Madison, WI, USA) according to the manufacturer's instructions. The cells were harvested and lysed in lysis buffer for 30 min with shaking for 10 s at 4 °C. After centrifugation, the supernatant was collected, and the protein concentration was determined. Each sample (50 μL) was incubated with caspase-3 or -9 substrate (5 μL) and 2×reaction buffer (50 μL) at 37 °C for 4 h. Colorimetric detection was performed at a wavelength of 405 nm.
ERK1/2 siRNA transfection
To silence the expression of ERK1/2 protein, SGC-7901 cells were transfected with a specific ERK1/2 siRNA. Non-targeted siRNA was used as a negative control. ERK1/2 siRNA and the control siRNA were transiently transfected into cells using the Lipofectamine 2000 transfection reagent according to the manufacturer's protocol. The efficiency of ERK1/2 protein silencing was analyzed by Western blot at 48 h.
Western blot analysis
Cell lysates were prepared with lysis buffer [1% Triton X-100, 150 mmol/L NaCl, 10 mmol/L Tris-HCl (pH 7.4), 1 mmol/L EDTA, 1 mmol/L EGTA (pH 8.0), 0.2 mmol/L Na3VO4, 0.2 mmol/L phenylmethylsulfonyl fluoride and 0.5% NP-40]. Equal amounts of protein (20–50 μg) were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and electrotransferred to polyvinylidene difluoride (PVDF) membranes. The membranes were blocked with 5% bovine serum albumin (BSA) for 2 h at room temperature and incubated overnight with specific antibodies as described above, followed by incubation with a horseradish peroxidase (HRP)-conjugated secondary antibody (1:10 000) for 2 h. The blots were evaluated with an enhanced chemiluminescence (ECL) detection system. Protein bands of interest were quantified using Quantity One software (Bio-Rad, Hercules, CA, USA), and the integrated signal densities were normalized to β-actin (loading control) and subsequently expressed in terms of the fractional abundance relative to control cells. These experiments were replicated three times.
Statistical analysis
Data from at least three independent experiments with duplicate determinations are expressed as the mean±SEM. Statistical analysis was performed using Student's t-test. The animal study was analyzed by one-way analysis of variance (ANOVA) followed by Tukey's test. Statistical significance was set at P<0.05.
Results
CP inhibits the growth of gastric cancer cells in vitro
Initially, we determined the effect of CP on the growth of various carcinoma cell lines using the MTT cellular survival assay. The results showed that CP treatment inhibited cellular growth with similar IC50 values (approximately 32–39 μmol/L) after a 24 h treatment period (Figure 1B). These findings indicate that CP is a broad-spectrum inhibitory agent of human carcinoma cells. Sensitivities to CP were assessed further in three different human gastric cancer cell lines, which were considered to be either well-differentiated (MKN-28), moderately differentiated (SGC-7901) or poorly differentiated (BGC-823). As shown in the growth curve (Figure 1C), CP treatment attenuated the growth rate of the three gastric cancer cell lines in a time-and dose-dependent manner. These results indicate that the cytotoxic effects of CP are not influenced by the differentiation status of the cells.
Furthermore, we assessed sensitivity to CP in normal cells. CP had insignificant effects on the viability of normal human gastric epithelial cells (GES-1) following 24 h of treatment at a concentration of 80 μmol/L (Figure 1C). Thus, the above results confirm that CP specifically inhibits cancer cell growth.
CP retards tumor growth in vivo
The in vivo anti-tumor effect of CP was evaluated by using SGC-7901-derived cancer xenografts in nude mice after 4 weeks of CP treatment. As shown in Figure 2 and Table 1, CP [200 μg/kg, intraperitoneally (ip)] caused significant inhibition of tumor growth, which was observed as early as 18 d after treatment and persisted after 30 d.

Anti-tumor effect of CP in vivo. Four-week-old female nude mice were injected subcutaneously (sc) with SGC-7901 cells in the left armpit region. When the mice produced palpable tumors, they were randomly assigned to groups for treatment with daily intraperitoneal (ip) injections of CP (200 μg/kg body weight) or vehicle (0.16% NaHCO3). Tumor volumes were measured using the formula mm3=width2×length/2. Mean±SEM. n=6. bP<0.05, cP<0.01 compared with the vehicle-treated group.
Effect of CP on cell cycle distribution
The results described above indicate that CP significantly inhibits the growth of gastric cancer cells. To determine whether the anti-tumor effects of CP were caused by cell cycle accumulation at a certain phase, we then analyzed the cell cycle population distribution in SGC-7901 cells. After treatment with 40 μmol/L CP for 0, 6, 12, and 24 h, the cells were stained with PI. PI-positive cells were detected by flow cytometric analysis. As shown in Figure 3A, treatment with CP led to the accumulation of cells in the G2/M phase. In parallel with the G2/M block, the cell cycle analysis showed a clear increase in the proportion of sub-G1 cells, which is regarded as a characteristic of apoptotic cells. Thus, these observations suggest that the inhibitory effect of CP on gastric cancer cells is, at least in part, due to G2/M arrest of the cell cycle.

Effects of CP on cell cycle distribution and apoptosis. SGC-7901 cells were treated with 40 μmol/L CP. (A) Cell cycle distribution was altered by CP treatment. (B) DNA fragmentation was evaluated using a Cell Death Detection ELISAPLUS Kit. The data are expressed as the mean±SEM of three separate experiments. bP<0.05, cP<0.01 compared with the group without CP treatment. (C) Annexin V/PI double-labeled flow cytometry assay. The percentage of annexin V-positive cells increased in a time-dependent manner (mean of three independent experiments) after CP treatment.
Effect of CP on gastric cancer cell apoptosis
The reduction in growth of gastric cancer cells in response to CP could be explained either by increased cell death or by reduced cell proliferation. SGC-7901 cells were treated with 40 μmol/L CP for 0, 6, 12, and 24 h, and apoptosis was assayed by Cell Death Detection ELISAPLUS. Nucleosome fragmentation (an indicator of apoptosis) confirmed that cells underwent apoptosis when treated with 40 μmol/L CP for 6 h, with the highest percentage of apoptotic cells observed at 24 h (Figure 3B). After exposure to 40 μmol/L CP for 0, 6, 12, and 24 h, flow cytometry using the FITC-annexin V/PI double staining method was used to generate an apoptotic cell scatterplot. The results showed that there was an increase in annexin V-positive cells after CP treatment (Figure 3C). Therefore, it is likely that CP treatment induced apoptosis but not necrosis in SGC-7901 cells.
Effects of CP on caspase activity and apoptosis protein expression in gastric cancer cells
The activation of caspases and cleavage of the nuclear protein PARP are also hallmarks of apoptosis18. PARP cleavage indicates caspase-3 activity and is used as a general marker for apoptosis. Our results showed that CP treatment increased cleaved caspase-3 and cleaved caspase-9 protein expression in SGC-7901 cells (Figure 4A). Additionally, caspase activity was measured using Caspase-Glo assays. As shown in Figure 4B, the activities of caspase-3 and -9 were significantly increased after CP treatment in SGC-7901 cells. The protein expression levels of cleaved PARP were examined by western blot after CP treatment (Figure 4C).

Effects of CP on caspase activity and apoptosis protein expression. After treatment with 40 μmol/L CP for the indicated times, SGC-7901 cells were harvested, and whole cell protein lysates were prepared. (A) Protein expression levels of cleaved caspase-3 and caspase-9 were measured by Western blot. (B) Changes in caspase activities induced by CP. (C) Cleavage of PARP induced by CP. (D) Bcl-2, Bax, and Bad protein expression was detected by Western blot. (E) Time-dependent changes in the Bax/Bcl-2 ratio after CP treatment. The data obtained from Western blot analysis of Bax and Bcl-2 were used to evaluate the effect of CP on the Bax/Bcl-2 ratio. The data are the mean±SEM of three separate experiments performed with similar results. bP<0.05, cP<0.01 compared with the group without CP treatment.
Because the Bcl-2 family members, including Bcl-2, Bcl-xL, Bad, and Bax, are recognized as important mediators in the apoptosis signaling pathway19, changes in Bcl-2, Bax, and Bad protein expression after CP treatment at various time points were investigated (Figure 4D). Marked increases in the levels of Bax and Bad began at 6 h and peaked at 24 h after CP treatment in SGC-7901 cells. In contrast, a reduction in Bcl-2 protein appeared later at 12 h. The ratio of Bax to Bcl-2 is the determining factor for the induction of apoptosis20. Densitometric analysis of Bax and Bcl-2 bands was performed using TotalLab TL120 software, and the data (relative density normalized to β-actin) were plotted as Bax/Bcl-2 ratios. The results in Figure 4E show that the Bax/Bcl-2 ratio gradually increased in a time-dependent manner.
CP induces the activation of ERK1/2 signaling in gastric cancer cells
To determine whether MAP kinase is involved in apoptosis in response to CP, we examined the effects of CP on the activation of several MAP kinase pathways. The phosphorylation of three MAPKs (ERK 1/2, JNK, and p38 MAPK) in response to CP treatment was analyzed via western blot using specific anti-phospho-kinase antibodies. As shown in Figure 5A, CP treatment induced the activation of ERK1/2 in a time-dependent manner. In contrast, the expression and phosphorylation levels of JNK and p38 were not altered after CP treatment (Figure 5B). Meanwhile, CP treatment induced the activation of MEK1/2 and raf-1 in a time-dependent manner (Figure 5C).

CP stimulates MAP kinase activation. After treatment with 40 μmol/L of CP or vehicle (0.16% NaHCO3), SGC-7901 cells were harvested at the indicated times. Protein expression of (A) ERK1/2 and phospho-ERK1/2, (B) p38 and JNK, and (C) MEK1/2 and Raf-1 was determined by Western blot analysis. β-Actin was used as a control to determine equal protein loading for each sample. The data are representative of three independent experiments.
Role of ERK1/2 in CP-induced caspase-9 activity and PARP cleavage
Furthermore, we examined the role of ERK1/2 in CP-induced apoptosis in SGC-7901 cells using an RNAi method and the ERK1/2 inhibitor PD98059. We observed that ERK1/2 siRNA-treated SGC-7901 cells showed reduced expression of ERK1/2 protein compared with those treated with the control siRNA (Figure 6A). As shown in Figure 6B, the ERK1/2 inhibitor PD98059 rescued the reduction of cell viability caused by CP treatment. Transfection of cells with the ERK1/2 siRNA also blocked the CP-induced loss of cell viability. These results suggest that the activation of ERK1/2 signaling is responsible for CP-induced apoptosis. These results were further confirmed by flow cytometric analysis, which was performed to determine the sub-G1 DNA content (Figure 6C). Similarly, the inhibition of ERK1/2 by its specific inhibitor or siRNA effectively downregulated CP-induced caspase-9 activity and PARP cleavage (Figure 6D). Moreover, the stimulatory effect of CP on ERK1/2 phosphorylation was significantly reduced by PD98059 (Figure 6E). The above results indicate that activation of ERK1/2 is important for CP-induced apoptosis.

Effect of ERK1/2 siRNA and an ERK1/2 inhibitor on CP-induced apoptosis. SGC-7901 cells were incubated with either CP (40 μmol/L) or PD98059 (20 μmol/L) for 24 h. (A) ERK1/2 siRNA (25 nmol/L) blocked the expression of ERK1/2 protein compared with control siRNA (25 nmol/L) in SGC-7901 cells. (B) After CP treatment, cell viability (assessed by the MTT assay) was decreased by the inhibition of ERK1/2. (C) Analysis of sub G1 DNA content by flow cytometry. (D) Determination of caspase-9 activity by Caspase-Glo assays and Western blot analysis of PARP cleavage. (E) Effect of an ERK1/2 inhibitor on CP-induced ERK1/2 activation. All values are presented as the mean±SEM from at least three separate experiments.
Discussion
BPs are commonly used to treat malignant and benign skeletal diseases characterized by excessive bone resorption. In particular, zoledronic acid has shown profound beneficial effects in patients with skeletal metastases21. BPs act through the induction of osteoclast apoptosis, likely by inhibiting the isoprenylation of proteins required for osteoclast survival22,23. Moreover, direct anti-tumor effects of BPs have been demonstrated in several cancer types, including multiple myeloma24, pancreatic cancer25, prostate cancer26 and breast cancer27. A growing body of evidence indicates that BPs have anti-tumorigenic capabilities, and several studies have shown positive outcomes with BP therapies with regard to tumor growth and metastatic activity for patients affected by diverse malignances7,28,29,30,31.
In this study, we synthesized a new BP derivative with improved potency and pharmacological characteristics. The highly active derivative, CP, showed obvious anti-proliferative activity against several human cancer cell types, which could be attributed to differences in its structure (ie, a nitrogen-based side chain with an additional adenine) (Figure 1A) compared with that of the parent molecule. Our study showed that CP inhibited the growth of gastric cancer cells in a time- and dose-dependent manner in vitro. We also found that CP retarded xenograft tumor growth in vivo.
Cell cycle control is the major regulatory mechanism of cell growth. Many cytotoxic agents and/or DNA damaging agents arrest the cell cycle at the G1, S, or G2/M phase before inducing apoptotic cell death32. It has been found that cell cycle arrest may result in apoptosis due to the existence of cell cycle checkpoints and feedback control mechanisms33. In the present study, the results of flow cytometric analysis show a clear accumulation of SGC-7901 cells at the G2/M phase after CP treatment.
Apoptosis, which is a complex multi-step process, is induced by the regulation of Bcl-2 family members, changes in the mitochondrial signaling pathway, caspase activation and many other processes. Bcl-2 family members and the caspase cascade are the key mediators in the apoptotic signal transduction pathway, and members of the Bcl-2 family are associated with mitochondrial membrane integrity34. Previous studies have indicated that if Bax homodimers predominate, cell death will occur; however, when Bcl-2 and Bax heterodimerization prevails, cells can survive35. Thus, the ratio of Bax to Bcl-2 may be a critical factor in the cellular threshold for apoptosis36. Our results showed a significant decrease of Bcl-2 and increase of Bax after CP treatment in SGC-7901 cells, thus shifting the Bax/Bcl-2 ratio in favor of apoptosis.
The caspase cascade plays a key role in apoptosis, and caspases have been referred to as the “central executioners of apoptosis”. Caspase-9, as an initiator caspase, is activated during cell apoptosis. The activated caspase-9 (cleaved caspase-9) goes on to activate caspase-7 and caspase-3 to evoke downstream apoptotic events. Caspase-3 is an “executioner” protein that can cleave specific substrates such as PARP37. The activation of caspase-3 results in cleavage of cytoskeletal and nuclear proteins and nucleosomal fragmentation of DNA38. In the current study, we found that CP treatment increased the activities of caspase-3 and -9, which provided evidence that CP-induced apoptosis in SGC-7901 cells is mediated by a caspase-dependent pathway. In this study, the finding that caspase-9 activation was accompanied by PARP cleavage indicated that caspase-9 might play a key role in CP-induced apoptosis in SGC-7901 cells. Thus, death receptor- or mitochondria-mediated activation of caspase may be a potential mechanism underlying CP-induced apoptosis in gastric cancer cells.
To further determine the mechanistic basis of the potent CP effects, we investigated the possible role of ERK1/2 signaling in CP-induced apoptosis in human gastric cancer cells. MAPKs, including ERK1/2, p38 MAPK, and SAPK/JNK, comprise a family of serine/threonine kinases. MAPKs are activated by a variety of stimuli, including growth factors, cytokines, hormones and mechanical forces, and play major roles in cellular responses including gene expression and cell proliferation, motility, survival, death and differentiation39. In particular, the importance of the ERK1/2 pathway has been implicated in cellular apoptosis induced by various cytotoxic compounds40,41. The pro-apoptotic function of the Ras/Raf/ERK1/2 pathway is well documented for apoptosis induced by DNA damaging agents, such as etoposide42 and doxorubicin43. Depending on the cell type and the nature of the injury, activation of the Ras/Raf/ERK1/2 pathway is associated with the intrinsic apoptotic pathway, which is characterized by the release of cytochrome c from mitochondria44 and the activation of the initiator caspase-945, or the extrinsic apoptotic pathway, which relies on the activation of the initiator caspase-845. It has been reported that ERK activation mediates cell cycle arrest and apoptosis after DNA damage43. Together, these observations suggest that the ERK1/2 signaling pathway may be an important target for CP in the treatment of gastric cancer cells. In the present study, CP induced the activation of ERK1/2 beginning 1 h after CP treatment, which was much earlier than the activation of apoptosis, and this activation was sustained for at least 24 h in SGC-7901 cells. We presumed that this elevation of ERK1/2 phosphorylation might also contribute to the activation of cellular apoptosis in vitro. Many studies have reported that activation of ERK1/2 by its upstream kinases or molecules is critical for its function. A number of factors, such as MAP kinase kinase 4 (MKK4), have been implicated in the activation of ERK1/2. In the present study, CP treatment induced the activation of MEK1/2 and raf-1, which are upstream of ERK1/2, in a time-dependent manner. In addition, our data showed that an ERK1/2-specific inhibitor or an ERK1/2 siRNA at least partially inhibited CP-induced apoptosis, indicating that ERK1/2 plays a key role in CP-initiated apoptosis. Although the results showed that the ERK1/2 pathway is necessary for CP-induced apoptosis in gastric cancer cells, inhibition of ERK1/2 using a specific inhibitor or siRNA did not result in total abolition of CP-induced cell death, as shown by MTT and flow cytometry assays. Thus, it is likely that ERK1/2-independent mechanisms participate in CP-induced apoptosis.
In conclusion, our results illustrate the effects of CP on SGC-7901 cells, and we found that CP inhibited cell growth by inducing apoptosis. The molecular mechanism of apoptosis involves the activation of the ERK1/2 signaling pathway, and Bcl-2 family members as well as the caspase cascade also contribute to apoptosis. Moreover, CP treatment caused significant inhibition of tumor growth in vivo. Therefore, CP may have potential for use in the treatment of gastric cancer.
Author contribution
Hai-jun WANG and Yu LIU performed the research and drafted the manuscript; Li-qiao FAN performed some of the experiments; Cai-li HAN, Ye JIANG, and Shu-jie CHENG performed the statistical analyses and helped draft the manuscript; Yong LI conceived the study and participated in its design and coordination.
References
Boissier S, Ferreras M, Peyruchaud O, Magnetto S, Ebetino FH, Colombel M, et al. Bisphosphonates inhibit breast and prostate carcinoma cell invasion, an early event in the formation of bone metastases. Cancer Res 2000; 60: 2949–54.
Van Pluijm G, Vloedgraven H, van Beek E, van Wee-Pals L, Löwik C, Papapoulos S . Bisphosphonates inhibit the adhesion of breast cancer cells to bone matrices in vitro. J Clin Invest 1996; 98: 698–705.
Santini D, Vincenzi B, Dicuonzo G, Avvisati G, Massacesi C, Battistoni F, et al. Zoledronic acid induces significant and long-lasting modifications of circulating angiogenic factors in cancer patients. Clin Cancer Res 2003; 9: 2893–7.
Fromigue O, Lagneaux L, Body JJ . Bisphosphonates induce breast cancer cell death in vitro. J Bone Miner Res 2000; 15: 2211–21.
Lowe LC, Senaratne SG, Colston KW . Induction of apoptosis in breast cancer cells by apomine is mediated by caspase and p38 mitogen activated protein kinase activation. Biochem Biophys Res Commun 2005; 329: 772–9.
Verdidk R, Franke HR, Wolbers F, Vermes I . Differential effects of bisphosphonates on breast cancer cell lines. Cancer Lett 2007; 246: 308–12.
Gnant M, Mlineritsch B, Schippinger W, Luschin-Ebengreuth G, Postlberger S, Menzel C, et al. Endocrine therapy plus zoledronic acid in premenopausal breast cancer. New Engl J Med 2009; 360: 679–91.
Toru H, Paul JW, Gregory RM, Toshiyuki Y . The bisphosphonate ibandronate promotes apoptosis in MDA-MB-231 human breast cancer cells in bone metastases. Cancer Res 2001; 61: 4418–24.
Kiagia M, Karapanagiotou E, Charpidou A, Dilana K, Dionellis G, Dannos I, et al. Rapid infusion of ibandronate in lung cancer patients with bone metastases. Anticancer Res 2006; 26: 3133–6.
Bergner R, Henrich DM, Hoffmann M, Honecker A, Mikus G, Nauth B, et al. Renal safety and pharmacokinetics of ibandronate in multiple myeloma patients with or without impaired renal function. J Clin Pharmacol 2007; 47: 942–50.
Thaler R, Spitzer S, Karlic H, Berger C, Klaushofer K, Varga F . Ibandronate increases the expression of the pro-apoptotic gene FAS by epigenetic mechanisms in tumor cells. Biochem Pharmacol 2013; 85: 173–85.
Lipton A, Cook RJ, Major P, Smith MR, Coleman R . Zoledronic acid and survival in breast cancer patients with bone metastases and elevated markers of osteoclast activity. Oncologist 2007; 12: 1035–43.
Oura S, Hirai I, Yoshimasu T, Kokawa Y, Sasaki R . Clinical efficacy of bisphosphonate therapy for bone metastasis from breast cancer. Breast Cancer 2003; 10: 28–32.
Yamada J, Hirokazu NT, Kitayama J, Yoneyama S, Tsuchiya T, Asakage M, et al. Zoledronic acid inhibits gastric cancer cells by induction of apoptosis. AACR Meeting Abstracts 2006; 4: 895.
Trojana J, Kimb SZ, Engelsc K, Kriener S, Mitroub PS, Chowb KU . In vitro chemosensitivity to gemcitabine, oxaliplatin and zoledronic acid predicts treatment response in metastatic gastric cancer. Anticancer Drugs 2005; 16: 87–91.
Ye J, Cai LH, Li GL, Tai MC, inventors; Shijiazhuang GUOYU patent trademark office LTD, assignee, A adenyl-diphosphonate preparation method and its application in pharmaceutic preparation. China patent 200710185250.7, 2008 Apr 9.
Liu ZF, Chen CY, Tang W, Zhang JY, Gong YQ, Jia JH . Gene-expression profiles in gastric epithelial cells stimulated with spiral and coccoid Helicobacter pylori. J Med Microbiol 2006; 55: 1009–15.
Jiang C, Wang Z, Ganther H, Lu J . Caspases as key executors of methyl selenium-induced apoptosis (anoikis) of DU-145 prostate cancer cells. Cancer Res 2001; 61: 3062–70.
Han LL, Xie LP, Li LH, Zhang XW, Zhang RQ, Wang HZ . Reactive oxygen species production and Bax/Bcl-2 regulation in honokiol-induced apoptosis in human hepatocellular carcinoma SMMC-7721 cells. Environ Toxicol Pharmacol 2009; 28: 97–103.
Cory S, Adams JM . The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer 2002; 2: 647–56.
Kohno N, Aogi K, Minami H, Nakamura S, Asaga T, Iino Y, et al. Zoledronic acid significantly reduces skeletal complications compared with placebo in Japanese women with bone metastases from breast cancer: a randomized, placebo-controlled trial. J Clin Oncol 2005; 23: 3314–21.
Luckman SP, Hughes DE, Coxon FP, Russell RG, Rogers MJ . Nitrogen-containing bisphosphonates inhibit the mevalonate pathway and prevent post-translational prenylation of GTP binding proteins, including Ras. J Bone Miner Res 1998; 13: 581–9.
Van Beek E, Pieterman E, Cohen L, Lowik C, Papoulos C . Nitrogen-containing bisphosphonates inhibit isopentyl pyrophosphate isomerase/farnesyl pyrophosphate synthase activity with relative potencies corresponding to their antiresorptive potencies in vitro and in vivo. Biochem Biophys Res Commun 1999; 255: 491–4.
Shipman CM, Rogers MJ, Apperley JF, Russell RG, Croucher PI . Bisphosphonates induce apoptosis in human myeloma cell lines: a novel anti-tumour activity. Br J Haematol 1997; 98: 665–72.
Tassone P, Tagliaferri P, Viscomi C, Palmieri C, Caraglia M, D'Alessandro A, et al. Zoledronic acid induces antiproliferative and apoptotic effects in human pancreatic cancer cells in vitro. Br J Cancer 2003; 88: 1971–8.
Lee MV, Fong EM, Singer FR, Guenette RS . Bisphosphonate treatment inhibits the growth of prostrate cancer cells. Cancer Res 2001; 61: 2602–8.
Senaratne SG, Pirianov G, Mansi JL, Arnett TR, Colston KW . Bisphosphonates induce apoptosis in human breast cancer cell lines. Br J Cancer 2000; 82: 1459–68.
Pazianas M, Abrahamsen B, Eiken PA, Eastell R, Russell RG . Reduced colon cancer incidence and mortality in postmenopausal women treated with an oral bisphosphonate — Danish National Register Based Cohort Study. Osteoporos Int 2012; 23: 2693–701.
Bundred N . Antiresorptive therapies in oncology and their effects on cancer progression. Cancer Treat Rev 2012; 38: 776–86.
Rodrigues P, Hering FO, Meller A . Adjuvant effect of IV clodronate on the delay of bone metastasis in high-risk prostate cancer patients: a prospective study. Cancer Res Treat 2011; 43: 231–5.
Daubine F, LE Bot R, Marquez M, Nilsson S, Schroder T, Holmberg AR . Treatment of bone metastasis in prostate cancer: efficacy of a novel polybisphosphonate. Anticancer Res 2011; 31: 4141–5.
Murray AW . Recycling the cell cycle: cyclins revisited. Cell 2004; 116: 221–34.
Pietenpol JA, Stewart ZA . Cell cycle checkpoint signaling: cell cycle arrest versus apoptosis. Toxicology 2002; 181–182: 475–81.
Adams JM, Cory S . The Bcl-2 protein family: arbiters of cell survival. Science 1998; 281: 1322–6.
Mohanty IR, Arya DS, Gupta SK . Withania somnifera provides cardioprotection and attenuates ischemia-reperfusion induced apoptosis. Clin Nutr 2008; 27: 635–42.
Tamm I, Schriever F, Dorken B . Apoptosis: implications of basic research for clinical oncology. Lancet Oncol 2001; 2: 33–42.
Nicholson DW, Thornberry NA . Apoptosis. Life and death decisions. Science 2003; 299: 214–5.
Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M . Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature 1995; 376: 37–43.
Chang L, Karin M . Mammalian MAP kinase signalling cascades. Nature 2001; 410: 37–40.
Prashant KM, Narayana K, Neha S, Pushkar S . Interplay between MEK-ERK signaling, cyclin D1, and cyclin-dependent kinase 5 regulates cell cycle reentry and apoptosis of neurons. Mol Biol Cell 2012; 23: 3722–30.
Chambon JP, Soule J, Pomies P, Fort P, Sahuquet A, Alexandre D, et al. Tail regression in ciona intestinalis (prochordate) involves a caspase-dependent apoptosis event associated with ERK activation. Development 2002; 129: 3105–14.
Stefanelli C, Tantini B, Fattori M, Stanic I, Pignatti C, Clo C, et al. Caspase activation in etoposide-treated fibroblasts is correlated to ERK phosphorylation and both events are blocked by polyamine depletion. FEBS Lett 2002; 527: 223–8.
Tang D, Wu D, Hirao A, Lahti JM, Liu L, Mazza B, et al. ERK activation mediates cell cycle arrest and apoptosis after DNA damage independently of p53. J Biol Chem 2002; 277: 12710–7.
Kim GS, Hong JS, Kim SW, Koh JM, An CS, Choi JY, et al. Leptin induces apoptosis via ERK/cPLA2/cytochrome c pathway in human bone marrow stromal cells. J Biol Chem 2003; 278: 21920–9.
Yang R, Piperdi S, Gorlick R . Activation of the RAF mitogen-activated protein extracellular signal-regulated kinase kinase extracellular signal-regulated kinase pathway mediates apoptosis induced by chelerythrine in osteosarcoma. Clin Cancer Res 2008; 14: 6396–404.
Acknowledgements
This research was supported by the National Natural Science Foundation of China (No 81072033).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wang, Hj., Liu, Y., Fan, Lq. et al. A new bisphosphonate derivative, CP, induces gastric cancer cell apoptosis via activation of the ERK1/2 signaling pathway. Acta Pharmacol Sin 34, 1535–1544 (2013). https://doi.org/10.1038/aps.2013.103
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2013.103
Keywords
This article is cited by
-
New Medical/Biologic Paradigms in the Treatment of Bone Tumors
Current Surgery Reports (2014)
