
Abstract
Aim:
To explore the function of the conserved aromatic cluster F2135.47, F3086.51, and F3096.52 in human β3 adrenergic receptor (hβ3AR).
Methods:
Point mutation technology was used to produce plasmid mutations of hβ3AR. HEK-293 cells were transiently co-transfected with the hβ3AR (wild-type or mutant) plasmids and luciferase reporter vector pCRE-luc. The expression levels of hβ3AR in the cells were determined by Western blot analysis. The constitutive signalling and the signalling induced by the β3AR selective agonist, BRL (BRL37344), were then evaluated. To further explore the interaction mechanism between BRL and β3AR, a three-dimensional complex model of β3AR and BRL was constructed by homology modelling and molecular docking.
Results:
For F3086.51, Ala and Leu substitution significantly decreased the constitutive activities of β3AR to approximately 10% of that for the wild-type receptor. However, both the potency and maximal efficacy were unchanged by Ala substitution. In the F3086.51L construct, the EC50 value manifested as a “right shift” of approximately two orders of magnitude with an increased Emax. Impressively, the molecular pharmacological phenotype was similar to the wild-type receptor for the introduction of Tyr at position 3086.51, though the EC50 value increased by approximately five-fold for the mutant. For F3096.52, the constitutive signalling for both F3096.52A and F3096.52L constructs were strongly impaired. In the F3096.52A construct, BRL-stimulated signalling showed a normal Emax but reduced potency. Leu substitution of F3096.52 reduced both the Emax and potency. When F3096.52 was mutated to Tyr, the constitutive activity was decreased approximately three-fold, and BRL-stimulated signalling was significantly impaired. Furthermore, the double mutant (F3086.51A_F3096.52A) caused the total loss of β3AR function. The predicted binding mode between β3AR and BRL revealed that both F3086.51 and F3096.52 were in the BRL binding pocket of β3AR, while F2135.47 and W3056.48 were distant from the binding site.
Conclusion:
These results revealed that aromatic residues, especially F3086.51 and F3096.52, play essential roles in the function of β3AR. Aromatic residues maintained the receptor in a partially activated state and significantly contributed to ligand binding. The results supported the common hypothesis that the aromatic cluster F[Y]5.47/F[Y]6.52/F[Y]6.51 conserved in class A G protein-coupled receptor (GPCR) plays an important role in the structural stability and activation of GPCRs.
Similar content being viewed by others

Introduction
G protein-coupled receptors (GPCRs), which are characterised as seven transmembrane (TM) helices, comprise a large superfamily of membrane receptors involved in a wide range of signalling transduction pathways, which are mainly activated by agonists. Their activation mechanisms have been studied for decades, and the results show that they can transit through inactive-state and several active-state conformations with or without ligands1,2. Although GPCRs are activated by ligands with different chemical natures, they are believed to share a common molecular activation mechanism3,4. A number of biochemical and biophysical approaches, including site-directed spin labelling and various fluorescent technologies, have been applied to study the activation-associated conformational changes of GPCRs5,6,7,8. These studies indicated that the movement of the intracellular segments of TMs, especially the change of the orientations of TM-III and TM-VI, is likely a key element in the activation of GPCRs. Alternative experimental approaches including metal-ion site or disulphide engineering revealed that the extracellular segments of the TMs appear to move in the opposite direction of the intracellular segments. Thus, a global toggle switch mechanism for GPCR activation was proposed, whereby the extracellular segment of TM-VI was bent towards TM-III while the intracellular part moved away from TM-III3,9.
At the cytoplasmic surface, the E/DRY motif on TM-III is conserved among all classes of GPCRs. These amino acids form a network of polar interactions that bridge TM-III and TM-VI and that stabilise the inactive-state conformation, which is called the 'ionic lock'10. For β2 adrenergic receptor (β2AR), mutations of these residues, which weakened the strength of the ionic lock, increased constitutive activity11. This interaction network has been observed in crystal structures of the dark-state rhodopsin12,13, and it was broken in several other GPCR crystal structures bound with antagonists, such as β1 adrenergic receptor (PDB code: 2VT414) and A2A adenosine receptor (PDB code: 3EML15). Microswitch during the process of GPCR activation has also been explored for decades. The W6.48 of the CWXP motif in GPCR, which is located at the bottom of the main ligand-binding pocket, is generally expected to function as a key microswitch in GPCR activation16,17. The W6.48 changes position and interaction partners during receptor activation17,18,19. Moreover, the aromatic cluster around W6.48, F[Y]5.47/F[Y]6.52/F[Y]6.51, is proposed to play a role in initiating the receptor activation20. However, the detailed functional mechanisms of these residues need further investigation.
There are three members of βARs (β1AR, β2AR, and β3AR) that belong to class A GPCRs. β3AR was originally discovered in the 1980s. Early studies identified that human β3AR was mainly expressed on the surface of both white and brown adipocytes, mediating metabolic effects such as lipolysis and thermogenesis21. More recent reports showed that β3AR is also an attractive target for drugs against overactive bladder22,23, anxiety and depressive disorders24,25. Understanding the mechanism of β3AR activation should facilitate rational drug discovery and the design of new types of β3AR agonists. In this study, we employed site-directed mutagenesis and molecular docking to explore the detailed function of the conserved aromatic cluster F2135.47/F3096.52/F3086.51 in β3AR.
Materials and methods
Reagents
Ham's F12 nutrient medium, G418 and pCDNA3.1 were purchased from Invitrogen (Carlsbad, CA, USA). The human pCDNA3.1-β3AR (pCDNA3.1-hβ3AR) plasmid was obtained from Missouri S&T cDNA Resource Center (Rolla, MO, USA). The pCRE-Luc plasmid containing four copies of a consensus CRE was obtained from Stratagene (La Jolla, CA, USA). CHO-K1 and HEK-293 cells were purchased from ATCC (Rockville, MD, USA). Recombinant cell line CHO-mock was constructed in-house (CHO-K1 cells were stably co-transfected with pCDNA3.1-mock plasmid and pCRE-luc plasmid). Fetal bovine serum was supplied by Hyclone (Logan, UT, USA). FuGENE6 Transfection Reagent was supplied by Roche (Indianapolis, IN, USA). The Steady-Glo Luciferase Assay System was obtained from Promega (Madison, WI, USA). β3AR selective agonist BRL37344 (BRL) was obtained from Tocris Bioscience (Bristol, UK). Phosphatase and protease inhibitor cocktail was obtained from Sigma (St Louis, MO, USA). PVDF membranes were obtained from Bio-Rad Laboratories (Richmond, CA, USA). Anti-β3AR antibody (c-20) was obtained from Santa Cruz (Santa Cruz, CA, USA). Anti-β-Actin antibody was obtained from Sigma (St Louis, MO, USA). HRP-conjugated secondary antibodies were obtained from Jackson ImmunoResearch Laboratories (West Grove, PA, USA). ECL substrate was obtained from Pierce (Rockford, IL, USA).
Mutant construction, cell culture and transfections
Human β3AR mutants were constructed by a PCR-based site-directed mutagenesis approach. The PCR products were digested with DpnI restriction enzyme and were then transformed into competent cells. All of the mutants were verified by DNA sequencing analysis. HEK-293 cells were plated at a density of approximately 20 000 cells/well in 96-well plates with 100 μL of high glucose Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum in a humidified 5% CO2/95% air atmosphere at 37 °C. After 24 h, the cells were co-transfected with pcDNA3.1-hβ3AR (wild-type or mutant) and the luciferase reporter plasmid pCRE-Luc using FuGENE6 Transfection Reagent according to the manufacturer's instructions.
CRE-luciferase activity test
After 24 h of transfection, an additional 100 μL of DMEM containing different concentrations of BRL37344 (BRL) was added. Then, the plate was incubated at 37 °C (5% CO2) for 3 h. The media were removed, and luciferase activities were detected using Steady-Glo Luciferase Assay System on the Flexstation III instrument (Molecular Devices, CA, USA).
Western blotting
Cells were harvested and homogenised in RIPA lysis buffer (50 mmol/L Tris, pH 7.4, 150 mmol/L NaCl, 1 mmol/L EDTA, 0.1% SDS, 1% Triton X-100, 1% sodium deoxycholate, 1 mmol/L PMSF) with a phosphatase and protease inhibitor cocktail. Proteins were separated by SDS-PAGE and transferred to PVDF membranes. Membranes were incubated with primary anti-β3AR antibody (1:200) and anti-β-Actin antibody (1:1000), followed by HRP-conjugated secondary antibodies (1:5000). The resulting immunoblots were visualised using ECL substrate.
Homology modelling and molecular docking
The construction of the homology model of β3AR (32-237, 282-361) was based on the structure of β2AR with InsightII26. GPCR has at least two functional states: inactive (R) and active (R*), which have different conformations. To obtain an active conformation of β3AR, the structure of β2AR binding with an agonist (PDB ID: 3P0G27) was used for our homology modelling, which was retrieved from the Protein Data Bank. On the basis of the sequence alignment result, the coordinates in conserved regions were assigned. The N and C terminal residues and loops were added using Loop_Search module and refined using Discover module. Disulfide bonds were defined between Cys110 and Cys196 and between Cys189 and Cys195. Finally, the whole structure model was minimised by SYBYL6.8 (Tripos Inc, St Louis, MO, USA). The stereochemical quality was assessed by Procheck28.
Molecular docking was performed using AutoDock4.229. To prepare both the receptor and BRL, all hydrogens were initially added, Gasteiger charges were computed, and the non-polar hydrogens were then merged with the corresponding non-polar atoms. The receptor was considered completely rigid for the docking study. The active site was defined by a grid box as large as 60×60×60 points with a grid spacing of 0.375Å using AutoGrid4. The box was centred on the centre of the Asp117 residue in the receptor. The docking parameters were prepared as follows: ga_pop_size, 150; ga_num_evals, 2500000; ga_num_generations, 27000; ga_run, 100 and rmstol, 2.0. The Lamarckian genetic algorithm was applied to account for protein-ligand interactions. Finally, the ligand conformation was selected considering both the predicted binding free energy and binding modes in the β3AR pocket.
Statistical analysis
EC50 values were determined by nonlinear regression using GraphPad Prism software (San Diego, CA, USA).
Results
The relationship between the expression level and the pharmacological properties of β3AR
To explore the influence of the protein expression level/transfection efficiency of plasmid DNAs on the detected pharmacological properties of β3AR, we adjusted the percentage of β3AR in DNA mixture (pCDNA3.1-hβ3AR and pCDNA3.1) to mimic the different expression level/transfection efficiency of the receptor. The constitutive activity (basal activity) and response stimulated by BRL were determined. The basal activity and maximal efficacy stimulated by BRL were both reduced less than 50%, and the EC50 value was reduced less than three-fold as the percentage of β3AR in the DNA mixture changed from 100% to 20% (Table 1). Therefore, in our experimental system, the expression level/transfection efficiency of β3AR or mutants did not significantly influence the pharmacological properties of β3AR.
Functional analysis of F3086.51 in β3AR
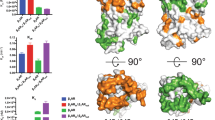
To validate the function of F3086.51, this aromatic residue was experimentally substituted with non-aromatic amino acid (Ala or Leu) and aromatic amino acid (Tyr), respectively. The results showed that all the mutants were well expressed in transfected HEK-293 cells as determined by Western blot analysis (the data for mutant F3086.51Y were not obtained) (Figure S1A). The activities of mutants were examined in transfected HEK-293 cells by measuring receptor-mediated luciferase activities. As was observed with substitution of F3086.51, both Ala and Leu substitution significantly decreased the constitutive activities of β3AR to approximately 10% of that for the wild-type receptor. However, both the potency and maximal efficacy were unchanged by Ala substitution. In the F3086.51L construct, the EC50 value manifested as a “right shift” of approximately two orders of magnitude with an increased Emax (Table 2, Figures 1A and 1B). Impressively, the molecular pharmacological phenotype was similar to the wild-type receptor for the introduction of Tyr at position 3086.51, though the EC50 value increased by approximately five-fold for the mutant (Table 2, Figure 1C). Thus, F3086.51 is essential for the constitutive signalling for the β3AR, and it could be involved in the activation process to some extent.

The pharmacological properties of HEK-293 cells transiently transfected with wild-type and mutated β3ARs (F3086.51A, F3086.51L, F3086.51Y, F3096.52A, F3096.52L, and F3096.52Y). The efficacy is detected as luciferase activity.
Functional analysis of F3096.52 in β3AR
F3096.52 was also substituted with non-aromatic amino acid (Ala or Leu) and aromatic amino acid (Tyr), respectively. According to the Western blot analysis results, Ala substitution reduced the expression of the receptor in transfected HEK-293 cells by approximately 25%, while the expression level of the Leu substituted mutant was increased by approximately 25% compared with the wild-type receptor (Figure S1). However, constitutive signalling for both F3096.52A and F3096.52L constructs were strongly impaired (Table 2, Figures 1D and 1E). In the F3096.52A construct, BRL-stimulated signalling showed a normal Emax but reduced potency (lgEC50 value is -6.6). Leu substitution of F3096.52 reduced both the Emax and potency (Table 2, Figures 1D and 1E). When F3096.52 was mutated to Tyr, the constitutive activity was decreased approximately three-fold, and BRL-stimulated signalling was significantly impaired (Table 2, Figure 1F). Thus, F3096.52 is essential for the constitutive signalling of β3AR and is important for the agonist-induced signalling.
To further explore the importance of F3086.51 and F3096.52, the double mutant F3086.51A_F3096.52A was constructed. The result showed that the protein expression level was reduced to approximately 30%, and both the constitutive and BRL-stimulated signalling were almost totally diminished (Table 2, Figures 2A and S1B).

The pharmacological properties of HEK-293 cells transiently transfected with wild-type and mutated β3ARs (F3086.51A_ F3096.52A, W3056.48A, F2135.47A). The efficacy is detected as luciferase activity.
When F2135.47 and W3056.48 were mutated to Ala, we observed that the constitutive activities and maximum responses (stimulated by BRL) were also dramatically impaired for both mutants (Table 2 and Figures 2B and 2C). The result was similar to that for the molecular pharmacological phenotype for the ghrelin receptor, a member of class A GPCR30.
The predicted binding model of β3AR and BRL
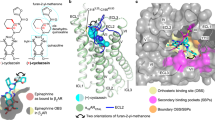
With the aim to explore the roles of the aromatic residues F2135.47/F3096.52/F3086.51 on the structural level, the interaction mechanism of β3AR and BRL was predicted by molecular docking. The 3D structure of β3AR was constructed by homology as described in the Materials and methods, and the stereochemical quality of the β3AR model was finally examined by PROCHECK. The result of PROCHECK showed that 94.4% of the residues were in the most favoured regions and that no residue was in the disallowed regions (Figure S2). BRL was then docked (via software AutoDock4.229) into the binding pocket of β3AR. As shown in Figure 3, BRL formed a hydrogen bond with residues Asp117, Ser208, and Asn312 (Figure 3A), which is consistent with the results reported for βAR family members31,32,33,34,35. F3086.51 and F3096.52 were in the binding pocket, and they had hydrophobic interactions with BRL, while F2135.47 was below the binding pocket (Figure 3A).

(A) The predicted binding mode of β3AR and BRL. (B) A π-π stacking is formed between F2135.47 and F3096.52 in the aromatic cluster F2135.47/F3096.52/F3086.51 in β3AR. Residues of β3AR are indicated in green sticks. BRL is shown in yellow sticks. The dashed lines in red represent hydrogen bonds.
A π-π stacking is formed between F2135.47 and F3096.52 in the aromatic cluster F2135.47/F3096.52/F3086.51 in β3AR (Figure 3B).
Discussion
The aromatic cluster F[Y]5.47/F[Y]6.52/F[Y]6.51 of GPCR is proposed to play a role in receptor activation20. F5.47 was found to serve as an aromatic lock for the proposed active conformation of the W6.48 “toggle switch” in seven-transmembrane receptor activation30. F6.52 was reported to be highly correlated with W6.48 and may mediate the TM6 Pro-kink19. In β3AR, the conserved aromatic cluster is F2135.47/F3096.52/F3086.51, and we studied the cluster's function on a cellular and structural level.
For GPCR assays, the most common cells used for generating cell lines are CHO-K1 and HEK-293 cells. With the aim to obtain a sensitive luminescent signal for our luciferase reporter gene system, three cell lines were chosen and compared: CHO-K1, HEK-293 and the in-house cell line CHO-mock. The results indicated that HEK-293 was the most suitable cell line for the transient expression of β3AR (Figure S3A and S3B). Hence, HEK-293 cells were transfected with wild-type or mutated β3AR in our study.
To explore the influence of transfection efficiency on the luciferase signal, different ratios of pCDNA3.1-hβ3AR plasmid to pCDNA3.1 plasmid were designed to mimic the different protein expression levels of β3AR. The results demonstrated that the expression level of β3AR did not greatly influence the luciferase signal in our experimental system (Table 1). In fact, this result was confirmed in our later Western bolt analysis results. For example, on residue F3096.52, Ala substitution reduced the expression of the receptor in transfected HEK-293 cells by approximately 25% and Leu substitution increased the expression approximately 25% compared to the wild-type receptor. In both cases, the constitutive signalling was strongly impaired (Table 2, Figures 1D and 1E).
From the results of the mutation of F3086.51 and F3096.52, and the double mutants, we discovered that both residues are essential for the constitutive activation of β3AR. Moreover, we showed that F3096.52 was not only essential for the constitutive signalling but also very important for the agonist-induced signalling. However, the results for F3086.51 demonstrated that this residue might not be critical for BRL-induced signalling efficacy, as both the potency and maximal efficacy were unaltered by Ala substitution (Table 2, Figures 1A). However, F3086.51 could be involved in the activation process to some extent.
Homology and molecular docking were carried out to further explore the role of the aromatic cluster on structural level. The predicted binding mode demonstrated that both F3086.51 and F3096.52 were located in the binding pocket and that they had hydrophobic interactions with BRL (Figure 3A), which are consistent with our results and the newly reported observations36. However, through the homology and molecular docking studies, we could not conclude that F3096.52 played a more important role than F3086.51, which did not correspond exactly to our experimental results. One possible reason for these results is that during the docking process, the conformation of β3AR was completely rigid; however, it has been reported that the conserved residue W3056.48 (W6.48) changes position during the receptor activation, which might drive the adjacent residues, F3086.51 and F3096.52 (Figure 3B), to undergo conformational changes19. Thus, rigid protein docking might not be accurate enough to predict the interaction mechanisms between ligands and GPCRs. Interestingly, as observed in Figure 3B, F2135.47 could form a π-π stacking with F3096.52. This interaction might have an essential role for both F2135.47 and F3096.52 in the signal transduction of β3AR.
As the aromatic cluster is highly conserved in GPCR, we carried out a statistic analysis throughout the class A GPCR. We surveyed all the members in the subfamily with the criterion of F[Y]5.47/F[Y]6.52/F[Y]6.51, as phenylalanine and tyrosine are highly homologous. The search results (Table S1) showed that the aromatic cluster resided in 18.5% of GPCR Class A members, and to be more precise, 72.1% of the amine subfamily, 75.7% of the rhodopsin subfamily, 91.3% of the gonadotropin-releasing hormone subfamily, and 18.4% of the peptide subfamily (the alignment and classification were taken from GPCRDB, http://www.gpcr.org/). Hence, the aromatic cluster is highly conserved in amine, rhodopsin, and gonadotropin-releasing hormone subfamilies and is important in a portion of the peptide subfamily members. In the class A GPCR, Phe or Tyr are conserved in the position of 5.47 (41.9%), 6.51 (75.6%), and 6.52 (75.1%, including β-ionone for rhodopsin).
In summary, we explored the importance of the aromatic residues near the binding pocket of β3AR by applying cell biology technologies and the molecular docking method. We discovered that the aromatic properties of F3086.51 and F3096.52 were essential in maintaining the receptor in a partially activated state as shown by the constitutive activity. Moreover, both of the residues contributed to hydrophobic interactions with BRL in binding with β3AR. In addition, the π-π stacking between F2135.47 and F3096.52 might explain their major roles in the signal transduction of β3AR. From a survey of the class A GPCR, we found that the aromatic cluster F[Y]5.47/F[Y]6.52/F[Y]6.51 was highly conserved, therefore, the aromatic cluster might have a common mechanism in GPCR activation.
Author contribution
Prof He-yao WANG and Prof Wei-liang ZHU designed and supervised the research and revised the manuscript; Hai-yan CAI performed the research, analysed data and wrote the manuscript; Prof Kai-xian CHEN helped with part of the research idea and helped revised the manuscript; Zhi-jian XU, Jie TANG, and Ying SUN helped with part of the research.
References
Kenakin T . Agonist-receptor efficacy I: mechanisms of efficacy and receptor promiscuity. Trends Pharmacol Sci 1995; 16: 188–92.
Lefkowitz RJ, Cotecchia S, Samama P, Costa T . Constitutive activity of receptors coupled to guanine nucleotide regulatory proteins. Trends Pharmacol Sci 1993; 14: 303–7.
Schwartz TW, Frimurer TM, Holst B, Rosenkilde MM, Elling CE . Molecular mechanism of 7TM receptor activation — a global toggle switch model. Annu Rev Pharmacol Toxicol 2006; 46: 481–519.
Schwartz TW, Rosenkilde MM . Is there a 'lock' for all agonist 'keys' in 7TM receptors? Trends Pharmacol Sci 1996; 17: 213–6.
Gether U, Lin S, Ghanouni P, Ballesteros JA, Weinstein H, Kobilka BK . Agonists induce conformational changes in transmembrane domains III and VI of the beta2 adrenoceptor. EMBO J 1997; 16: 6737–47.
Gether U, Lin S, Kobilka BK . Fluorescent labeling of purified beta 2 adrenergic receptor. Evidence for ligand-specific conformational changes. J Biol Chem 1995; 270: 28268–75.
Sheikh SP, Zvyaga TA, Lichtarge O, Sakmar TP, Bourne HR . Rhodopsin activation blocked by metal-ion-binding sites linking transmembrane helices C and F. Nature 1996; 383: 347–50.
Ghanouni P, Steenhuis JJ, Farrens DL, Kobilka BK . Agonist-induced conformational changes in the G-protein-coupling domain of the beta 2 adrenergic receptor. Proc Natl Acad Sci U S A 2001; 98: 5997–6002.
Elling CE, Frimurer TM, Gerlach LO, Jorgensen R, Holst B, Schwartz TW . Metal ion site engineering indicates a global toggle switch model for seven-transmembrane receptor activation. J Biol Chem 2006; 281: 17337–46.
Vogel R, Mahalingam M, Ludeke S, Huber T, Siebert F, Sakmar TP . Functional role of the “ionic lock” — an interhelical hydrogen-bond network in family A heptahelical receptors. J Mol Biol 2008; 380: 648–55.
Rasmussen SG, Jensen AD, Liapakis G, Ghanouni P, Javitch JA, Gether U . Mutation of a highly conserved aspartic acid in the beta2 adrenergic receptor: constitutive activation, structural instability, and conformational rearrangement of transmembrane segment 6. Mol Pharmacol 1999; 56: 175–84.
Li J, Edwards PC, Burghammer M, Villa C, Schertler GF . Structure of bovine rhodopsin in a trigonal crystal form. J Mol Biol 2004; 343: 1409–38.
Okada T, Sugihara M, Bondar A-N, Elstner M, Entel P, Buss V . The retinal conformation and its environment in rhodopsin in light of a new 2.2Å crystal structure. J Mol Biol 2004; 342: 571–83.
Warne T, Serrano-Vega MJ, Baker JG, Moukhametzianov R, Edwards PC, Henderson R, et al. Structure of a beta1-adrenergic G-protein-coupled receptor. Nature 2008; 454: 486–91.
Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EY, Lane JR, et al. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 2008; 322: 1211–7.
Visiers I, Ballesteros JA, Weinstein H . Three-dimensional representations of G protein-coupled receptor structures and mechanisms. Methods Enzymol 2002; 343: 329–71.
Klein-Seetharaman J, Yanamala NV, Javeed F, Reeves PJ, Getmanova EV, Loewen MC, et al. Differential dynamics in the G protein-coupled receptor rhodopsin revealed by solution NMR. Proc Natl Acad Sci U S A 2004; 101: 3409–13.
Crocker E, Eilers M, Ahuja S, Hornak V, Hirshfeld A, Sheves M, et al. Location of Trp265 in metarhodopsin II: implications for the activation mechanism of the visual receptor rhodopsin. J Mol Biol 2006; 357: 163–72.
Shi L, Liapakis G, Xu R, Guarnieri F, Ballesteros JA, Javitch JA . Beta2 adrenergic receptor activation. Modulation of the proline kink in transmembrane 6 by a rotamer toggle switch. J Biol Chem 2002; 277: 40989–96.
Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science 2007; 318: 1258–65.
Emorine LJ, Marullo S, Briend-Sutren MM, Patey G, Tate K, Delavier-Klutchko C, et al. Molecular characterization of the human beta 3-adrenergic receptor. Science 1989; 245: 1118–21.
Yamazaki Y, Takeda H, Akahane M, Igawa Y, Nishizawa O, Ajisawa Y . Species differences in the distribution of beta-adrenoceptor subtypes in bladder smooth muscle. Br J Pharmacol 1998; 124: 593–9.
Fujimura T, Tamura K, Tsutsumi T, Yamamoto T, Nakamura K, Koibuchi Y, et al. Expression and possible functional role of the beta3-adrenoceptor in human and rat detrusor muscle. J Urol 1999; 161: 680–5.
Lenard NR, Gettys TW, Dunn AJ . Activation of beta2- and beta3-adrenergic receptors increases brain tryptophan. J Pharmacol Exp Ther 2003; 305: 653–9.
Claustre Y, Leonetti M, Santucci V, Bougault I, Desvignes C, Rouquier L, et al. Effects of the beta3-adrenoceptor (Adrb3) agonist SR58611A (amibegron) on serotonergic and noradrenergic transmission in the rodent: relevance to its antidepressant/anxiolytic-like profile. Neuroscience 2008; 156: 353–64.
Accelrys. InsightII, Version 2005, Molecular modeling package. San Diego, CA: Molecular Simulation, Accelrys; 2005.
Rasmussen SG, Choi HJ, Fung JJ, Pardon E, Casarosa P, Chae PS, et al. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature 2011; 469: 175–80.
Laskowski RA, Macarthur MW, Moss DS, Thornton JM . Procheck — a program to check the stereochemical quality of protein structures. J Appl Crystallogr 1993; 26: 283–91.
Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, et al. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem 1998; 19: 1639–62.
Holst B, Nygaard R, Valentin-Hansen L, Bach A, Engelstoft MS, Petersen PS, et al. A conserved aromatic lock for the tryptophan rotameric switch in TM-VI of seven-transmembrane receptors. J Biol Chem 2010; 285: 3973–85.
Strader CD, Candelore MR, Hill WS, Dixon RA, Sigal IS . A single amino acid substitution in the beta-adrenergic receptor promotes partial agonist activity from antagonists. J Biol Chem 1989; 264: 16470–7.
Wong SK, Slaughter C, Ruoho AE, Ross EM . The catecholamine binding site of the beta-adrenergic receptor is formed by juxtaposed membrane-spanning domains. J Biol Chem 1988; 263: 7925–8.
Strader CD, Candelore MR, Hill WS, Sigal IS, Dixon RA . Identification of two serine residues involved in agonist activation of the beta-adrenergic receptor. J Biol Chem 1989; 264: 13572–8.
Takayuki S, Hiroyuki K, Taku N, Hitoshi K . Ser203 as well as Ser204 and Ser207 in fifth transmembrane domain of the human β2-adrenoceptor contributes to agonist binding and receptor activation. Br J Pharmacol 1999; 128: 272–4.
Wieland K, Zuurmond HM, Krasel C, Ijzerman AP, Lohse MJ . Involvement of Asn-293 in stereospecific agonist recognition and in activation of the β2-adrenergic receptor. Proc Natl Acad Sci U S A 1996; 93: 9276–81.
Jin F, Lu C, Sun X, Li W, Liu G, Tang Y . Insights into the binding modes of human beta-adrenergic receptor agonists with ligand-based and receptor-based methods. Mol Divers 2011; 15: 817–31.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (20721003, 81072681), the International S&T Cooperation(2010DFB73280), and the Shanghai Committee of Science and Technology International Cooperation Project (09540703900).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Supplementary figures are available at website of Acta Pharmacologica Sinica on NPG.
Supplementary information
Supplementary information, Figure S1
Expression levels of beta3AR mutants determined by Western bolt. (DOC 879 kb)
Supplementary information, Figure S2
Ramachandran plot for analyzing the rationality of beta3AR structure (DOC 230 kb)
Supplementary information, Figure S3
Cell lines comparison. (DOC 53 kb)
Supplementary information, Table S1
The amino acid distribution in position of 5.47, 6.51 and 6.52 in the Rhodopsin receptor family (class A) (DOC 38 kb)
Rights and permissions
About this article
Cite this article
Cai, Hy., Xu, Zj., Tang, J. et al. The essential role for aromatic cluster in the β3 adrenergic receptor. Acta Pharmacol Sin 33, 1062–1068 (2012). https://doi.org/10.1038/aps.2012.55
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2012.55
Keywords
This article is cited by
-
β3-adrenergic receptor activity modulates melanoma cell proliferation and survival through nitric oxide signaling
Naunyn-Schmiedeberg's Archives of Pharmacology (2014)
-
Functional involvement of β3-adrenergic receptors in melanoma growth and vascularization
Journal of Molecular Medicine (2013)
