
Abstract
Aim:
To characterize the pharmacokinetics (PKs), pharmacodynamics (PDs), and tolerability of different dose regimens of prasugrel in healthy Chinese subjects.
Methods:
This was a single-centered, open-label, parallel-design study. Subjects received a single loading dose (LD) of prasugrel followed by once-daily maintenance dose (MD) for 10 d. They were enrolled into 3 groups: 60 mg LD/10 mg MD; 30 mg LD/7.5 mg MD; 30 mg LD/5 mg MD. Blood samples were collected after the first and last dose. The serum concentration of the active metabolite of prasugrel was determined using a LC/MS/MS method. Platelet aggregation was assessed using the VerifyNow-P2Y12 assay.
Results:
Thirty-six healthy native Chinese subjects (19 males) aged 18–45 were enrolled; mean age and body weight were similar across the treatment groups (n=12 for each). The metabolite AUC0–4 and Cmax increased dose-proportionally across the dose range of 5 mg to 60 mg. The median Tmax was 0.5 h in all groups. The PD parameters, indicated by the inhibition of ADP-induced platelet aggregation, were met more rapidly in the 60 mg group than the 30 mg group after the LD (94%–98%). This high degree of inhibition of platelet aggregation was maintained following the 10 mg MD (87%–90%) and was lower in the 7.5 mg and 5 mg MD groups (79%–83% and 64%–67%, respectively). Prasugrel was well tolerated in healthy Chinese subjects for single doses up to 60 mg and a MD of 10 mg for 10 d.
Conclusion:
The PKs and PDs of the active metabolite of prasugrel were similar to those in Chinese subjects reported by a previous bridging study, which demonstrated that the exposure to the active metabolite in Chinese subjects was higher than in Caucasians.
Similar content being viewed by others


Introduction
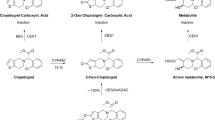
Platelets play an important role in atherothrombosis, and antiplatelet therapy is widely used in the prevention of atherothrombotic events. Prasugrel is a third-generation thienopyridine agent that was approved in the European Union, the United States and other regions in 2009 for the treatment of acute coronary syndrome (ACS) in patients undergoing percutaneous coronary intervention (PCI)1. To date, prasugrel has not been marketed in China. Prasugrel is a thienopyridine adenosine diphosphate (ADP) receptor antagonist prodrug that rapidly converts to an inactive metabolite (R-95913) by carboxyesterase and cannot be detected in plasma. The conversion of R-95913 to R-138727 is catalyzed by cytochrome P450 enzymes (Figure 1); R-138727 binds specifically and irreversibly to the P2Y12 ADP receptor and inhibits platelet activation and aggregation for the remainder of the life of the platelet2.

Structure and primary metabolic pathways of prasugrel.
Compared with clopidogrel in a phase III trial (TRITON-TIMI 38), 13608 patients with moderate- to high-risk ACS undergoing scheduled PCI after having taken an aspirin regimen received prasugrel or clopidogrel for 6 to 15 months. Prasugrel therapy was associated with significantly reduced rates of ischemic events, including stent thrombosis, but with an increased risk of major bleeding3.
Prasugrel's pharmacokinetics (PKs) are similar in healthy subjects, patients with stable atherosclerosis and patients undergoing PCI. After a loading dose (LD) of 60 mg, the active metabolite (Pras-AM) appears quickly in plasma; Tmax occurs at approximately 30 min, with terminal elimination T1/2 occurring at approximately 7.4 h. The apparent CL of Pras-AM is 149 L/h, and the apparent Vd is 66.4 L4.
Earlier studies were conducted primarily in Caucasian groups, and the dosing regimen was a 60 mg LD and a 10 mg maintenance dose (MD). Studies on healthy Caucasian and Chinese subjects suggested that Pras-AM exposure was higher in Chinese subjects than that in Caucasians5; the study in Chinese, Korean, and Japanese populations also showed higher exposure to Pras-AM and higher degree of platelet inhibition in these groups than in Caucasian populations6.
Considering the PKs and pharmacodynamics (PDs) of drug exposure, to reduce the risk of bleeding and other adverse events, a lower dose regimen may be more favorable for the Chinese population. Because the data on the Chinese population were obtained from subjects outside of China, information regarding prasugrel exposure in native Chinese subjects is limited. The dose regimen we designed for healthy Chinese subjects included a standard regimen of a 60 mg LD with a 10 mg MD and a 30 mg LD with a 7.5 mg MD and a 5 mg MD.
Materials and methods
The study was conducted in accordance with the Declaration of Helsinki (World Medical Association), Good Clinical Practice (GCP) guidelines, and the laws and regulations of China. The study protocol and informed consent forms were approved by the Independent Ethics Committee and the Institutional Review Board of Peking University First Hospital and the State Food and Drug Administration (SFDA) of China under SFDA approval Nos 2009L01051 (5 mg), 2009L01052 (7.5 mg), and 2009L01053 (10 mg). Prior to the beginning of the study, all of the subjects provided written informed consent.
Subjects
Healthy volunteer male and female Chinese subjects between the ages of 18 and 45 with a body mass index (BMI) of 19 kg/m2 to 24 kg/m2 were included in the study. Eligibility was based on medical history, physical examination, clinical laboratory tests, and an electrocardiogram (ECG). The exclusion criteria included a history of coagulation or bleeding disorders, a platelet count of <100 000 cell/mm3, and other abnormal coagulation test results at screening. The use of prescribed, over-the-counter (other than thyroid medication, oral contraceptive treatments, and hormone replacement therapy), or herbal medications (other than vitamin/mineral supplements) was not permitted within 14 d prior to study dosing. Because prasugrel is an antiplatelet drug, the use of aspirin, other non-steroidal anti-inflammatory drugs, and other drugs known to affect platelet function was not permitted within 14 d prior to dosing or during the study.
Study design and drug administration
This was a single-center, open-label, parallel-design study conducted from July 2009 to September 2009 at Peking University First Hospital in China. The study was sponsored by Eli Lilly and Company, Indianapolis, IN, USA.
The subjects were randomized to 1 of 3 prasugrel dosing groups. The LD (dose received on d 1) was 60, 30, or 30 mg, and the MD (dose received once daily on d 2 through 11) was 10, 7.5, or 5 mg. The dosing regimens were based on other trials that identified higher overall exposure and an elevated overall PD response in Asian versus Caucasian subjects in addition to subgroup analyses by ethnicity, body weight, and age5,6,7,. The drug was supplied as a tablet containing 5, 7.5, or 10 mg of prasugrel as a hydrochloride salt.
The subjects entered the clinical research unit (CRU) the evening before the initial (d 1) and final (d 11) dosing, resided there for approximately 24 h post-dose, and returned for daily dosing on the mornings of d 2 through 10. The subjects fasted overnight prior to taking prasugrel and continued to fast for up to 2 h post-dose. The subjects returned to the CRU for a follow-up visit between 10 and 30 d after the last prasugrel dose.
Safety data
Safety was assessed subjectively and objectively, and the data collected during the study included the occurrence of adverse events (AEs), clinical laboratory tests, vital signs, 12-lead ECG, physical examinations, and petechiae examination. Symptoms were classified using the Medical Dictionary for Regulatory Activities.
Pharmacokinetic sample collection
Three-milliliter blood samples were collected at 0.25, 0.5, 1, 2, 4, 6, 8, 10, 12, and 24 h post-dose on d 1 and d 11 for the measurement of Pras-AM (R-138727) serum concentrations.
Pharmacokinetic sample preparation
The pharmacokinetic blood samples were reacted with 25 μL of 500 mmol/L 3'-methoxyphenacyl bromide in acetonitrile within 30 s of collection to derivatize and stabilize the active metabolite. The separated serum samples were stored at approximately -70 °C in labeled polypropylene tubes until they were shipped on dry ice to Advion Biosciences, Ithaca, NY, USA, for analysis.
Analytical assays
The serum samples were analyzed for Pras-AM at Advion Biosciences using a validated liquid chromatography with tandem mass spectrometric detection (LC/MS/MS) method8. Originally validated for plasma samples before the study, the method was confirmed to also be suitable for serum. The lower and upper limits of quantification were 0.5 ng/mL and 250 ng/mL, respectively. Samples above the limit of quantification were diluted and reanalyzed to yield results within the calibrated range.
Pharmacodynamic evaluations
Blood samples of approximately 2 mL were collected into citrate tubes for the assessment of platelet aggregation using a VerifyNow-P2Y12 (VN-P2Y12) assay (Accumetrics, San Diego, CA, USA). Samples were collected at d 1 pre-dose; 0.5, 1, 2, 4, and 24 h post-dose; on d 10 pre-dose; and on d 11 pre-dose and 24 h post-dose. The data were recorded directly from the VerifyNow device as percent inhibition of P2Y12 reaction units (%IPRU).
Pharmacokinetic parameters
The PK parameters for Pras-AM assessed included the area under the concentration-time curve (AUC), which was calculated through the sampling time of the last quantifiable serum concentration (AUC0–tlast), and the maximum observed serum concentration (Cmax). The time at which Cmax occurred (Tmax) was measured using non-compartmental methods. The AUC0–tlast and Cmax for Pras-AM were estimated at each dose level to delineate the effects of the different dosing regimens.
Pharmacodynamic parameters
The pharmacodynamic parameters reported from the VN-P2Y12 assay were summarized by dose group using distributive statistics.
Statistical analysis
Up to 40 subjects were to be enrolled in the trial so that at least 10 subjects could complete each of the 3 dosing regimens of the study. The sample size was determined according to the China Regulatory Authority recommendations that approximately 8 to 12 subjects be evaluated for each dosing group. All of the statistical analyses were carried out using SAS software (Version 9.1, SAS Institute Inc, Cary, NC, USA).
A power model was used to estimate the dose proportionality; the log-transformed dose was fitted as the fixed effect, and subjects were fitted as the random effect9. The log-transformed AUC through 4 h (AUC0–4) and Cmax were analyzed separately. AUC0–4 was used because it represented the interval over which the active metabolite concentrations were quantifiable in all subjects at all dose levels, thereby eliminating the confounding influence of the quantitation limit on the AUC estimation.
Results
Demographic characteristics
Thirty-six healthy, native Chinese subjects (19 males, 17 females) were enrolled, and 35 subjects completed the study. The mean age, body weight, and BMI were similar across the treatment groups (Table 1).
Safety and tolerability
The safety data included all 36 subjects, who each received at least one dose of prasugrel. LDs of 60 mg and 30 mg and MDs of 10, 7.5, and 5 mg were well tolerated. No AEs were reported in the 30 mg/5 mg group, and the incidence of AEs in the 30 mg/7.5 mg and 60 mg/10 mg groups was similar and low. No serious AEs were reported in any group, and all of the AEs were mild and resolved without treatment. The most common AE was ecchymosis, but the frequency was low (only 8% of subjects). The only AE associated with bleeding was the presence of red blood cells in the urine, which lasts for 5 d after first dosing without complaint; it was typically resolved without treatment and was considered to be mild in severity. Because the red blood cells were only detected with a low power microscope, this AE is not considered be clinically significant. Only one subject withdrew from the trial because of a rash, which appeared 2 h after the administration of a 30 mg LD; this AE was mild and resolved without treatment after approximately 2 d. The investigator considered the AE to be related to the treatment.
Pharmacokinetics
All 36 subjects enrolled in the trials contributed to the PK evaluation. The PK parameters are shown in Table 2, and the mean Pras-AM concentration versus time after the LD and MDs is shown in Figures 2A and 2B. The median Tmax value was 0.5 h in all of the dosing groups (from 0.25 to 1 h). The active metabolite Pras-AM, AUC0–tlast, AUC0–4, and Cmax increased proportionally to the dose of prasugrel from 5 mg to 60 mg. The beta values (95%CIs) for the Cmax and AUC0–4 were 1.064 (0.978, 1.148) and 1.113 (1.042, 1.186), respectively.

Serum concentration-time profiles of Pras-AM following different doses of prasugrel. (A) Serum concentrations after prasugrel LD of 60 mg and 30 mg on d 1. (B) Serum concentrations following prasugrel MD of 10, 7.5, and 5 mg on d 11. Data are presented as mean±SD. n=12. LD, loading dose; MD, maintenance dose.
The exponent for the power model equation for Pras-AM versus the prasugrel dose was close to 1; the equation showed that the AUC increased in proportion to dose across a range of 5 mg to 60 mg (Figure 3).

Individual estimates of the area under the serum concentration-time curve from time of dosing through 4 h postdose for prasugrel active metabolite, Pras-AM, across a dose range of 5 mg to 60 mg of prasugrel. AUC0–4, area under the curve through 4 h postdose.
Pharmacodynamics
The PD parameters were directly assessed using an Accumetrics VN-P2Y12 assay, and the maximum inhibition of ADP-induced platelet aggregation was similar after a 30 mg LD and a 60 mg LD. Aggregation occurred rapidly in the first hour of dosing at all the dose regimens but most rapidly at a 60 mg dose. The minimum IPRU values were observed at 4 h after each LD. The high degree of inhibition lasted longer in the 10 mg MD group but was slightly lower in the 5 mg and 7.5 mg MD groups. Compared with the LD, the reduction in PRU was greater in the 10 mg MD group and less pronounced in the 5 mg MD group. The mean device-reported inhibition was 64% to 67%, 79% to 83%, and 87% to 90% for the 5, 7.5, and 10 mg MD groups, respectively (Figure 4).

Arithmetic mean VerifyNow P2Y12 (VN-P2Y12) percent inhibition of P2Y12 reaction units (%IPRU) following a prasugrel loading dose and during prasugrel maintenance dose (MD).
Discussion
This study is the first to describe the PKs, PDs, and safety response of prasugrel in healthy native Chinese subjects. Prasugrel was well-tolerated with a low number of drug-related AEs.
Prasugrel was rapidly absorbed, with the Cmax for the active metabolite occurring 0.5 h post-dose. This rapid absorption is similar to previous observations in Caucasian and Eastern Asian groups showing that the Cmax was reached within 30 min. The PK parameters were different in the Caucasian groups, indicating that Chinese subjects experience higher exposure to the active metabolite, particularly when considering the AUC and the Cmax. Table 2 below shows a comparison of the essential PK parameters in this study and in a previously published bridging study, which showed similar PK parameters6. Prasugrel's exposure, in terms of AUC and Cmax, increased dose-dependently for doses from 5 mg to 60 mg.
The PD parameters assessed by the VN-P2Y12 assay showed that the inhibition of ADP-induced platelet aggregation occurred rapidly, within 1 h after dosing; the maximum PD effect was similar in the 60 mg and 30 mg LD groups, approximately 94%–98%. This high degree of inhibition of platelet aggregation was maintained during the steady states on d 10 and d 11 post-dose. The IPA of the 10 mg MD group reached 87%–90%, and that of the MD 7.5 and 5 mg groups was lower, at 79%–83% and 64%–67%, respectively. Compared with the previous study of Caucasian subjects, this effect was greater in Chinese subjects given the same dose of prasugrel.
This study clearly showed that prasugrel is well tolerated, with predictable dose-proportional PKs and dose-dependent anticoagulant activity in healthy Chinese subjects, for single doses ranging from 5 mg to 60 mg and daily MDs of up to 10 mg.
For all dose regimens administered in this study to healthy subjects, the AEs reported were all mild in severity, and no serious AEs were reported. There were no safety concerns reported in the clinical laboratory evaluations, vital signs, and ECGs following multiple doses of prasugrel.
Following administration, the exposure to prasugrel's active metabolite following administration was higher in Asian groups than in previously reported studies comparing Asian and Caucasian groups5,6. The lower mean body weight of Asian subjects compared to Caucasian subjects may contribute to the higher exposure of prasugrel's active metabolite in Asians. Variations in CYP2C19 and CYP2C9 genetics cannot explain the higher active metabolite exposure in Asians because CYP2C19 and CYP2C9 genotypes associated with reduced function do not appear to affect the prasugrel PK or PD response10.
Author contribution
Yi-min CUI, Zi-ning WANG, Hui-lin ZHANG, Xia ZHAO, and Ying ZHOU were investigators in this study, and Xiao-wen CHEN is an employee and stockholder of Eli Lilly and Company.
References
Baker WL, White CM . Role of prasugrel, a novel P2Y12 receptor antagonist, in the management of acute coronary syndromes. Am J Cardiovasc Drugs 2009; 9: 213–29.
Niitsu Y, Jakubowski JA, Sugidachi A, Asai F . Pharmacology of CS-747 (prasugrel, LY640315), a novel, potent antiplatelet agent with in vivo P2Y12 receptor antagonist activity. Semin Thromb Hemost 2005; 31: 184–94.
Wiviott SD, Braunwald E, McCabe CH, Montalescot G, Ruzyllo W, Gottlieb S, et al. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2007; 357: 2001–15.
Ernest CS II, Small DS, Rohatagi S, Salazar DE, Wallentin L, Winters KJ, et al. Population pharmacokinetics and pharamcodynamics of prasugrel and clopidogrel in aspirin-treated patients with stable coronary artery disease. J Pharmacokinet Pharmacodyn 2008; 35: 593–618.
Small DS, Payne CD, Kothare P, Yuen E, Natanegara F, Teng Loh M, et al. Pharmacodynamics and pharmacokinetics of single doses of prasugrel 30 mg and clopidogrel 300 mg in healthy Chinese and white volunteers: an open-label trial. Clin Ther 2010; 32: 365–79.
Small DS, Kothare P, Yuen E, Lachno DR, Li YG, Winters KJ, et al. The pharmacokinetics and pharmacodynamics of prasugrel in healthy Chinese, Japanese, and Korean subjects compared with healthy Caucasian subjects. Eur J Clin Pharmacol 2010; 66: 127–35.
Small DS, Li YG, Ernest CS II, April JH, Farid NA, Payne CD, et al. Integrated analysis of pharmacokinetic data across multiple clinical pharmacology studies of prasugrel, a new thienopyridine antiplatelet agent. J Clin Pharmacol 2011; 51: 321–32.
Farid NA, McIntosh M, Garofolo F, Wong E, Shwajch A, Kennedy M, et al. Determination of the active and inactive metabolites of prasugrel in human plasma by liquid chromatography/tandem mass spectrometry. Rapid Commun Mass Spectrom 2007; 21: 169–79.
Smith BP, Vandenhende FR, DeSante KA, Farid NA, Welch PA, Callaghan JT, et al. Confidence interval criteria for assessment of dose proportionality. Pharm Res 2000; 17: 1278–83.
Brandt JT, Close SL, Iturria SJ, Payne CD, Farid NA, Ernest CS II, et al. Common polymorphism of CYP2C19 and CYP2C9 affect the pharmacokinetic and pharmacodynamic response to clopidogrel but not prasugrel. J Thromb Haemost 2007; 5: 2429–36.
Acknowledgements
The study was financially supported by Eli Lilly and Company, Indianapolis, Indiana, USA. The authors are indebted to the patients who participated in this study and their families and to all of the physicians, nurses, and study coordinators who cared for the patients. The authors acknowledge the following persons for their assistance in the study and the development of the manuscript: Pei-hong SUN, Yan LIANG, Dong-mei ZHANG and Jing-zi LI.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Cui, Ym., Wang, Zn., Chen, Xw. et al. Pharmacokinetics and pharmacodynamics of single and multiple doses of prasugrel in healthy native Chinese subjects. Acta Pharmacol Sin 33, 1395–1400 (2012). https://doi.org/10.1038/aps.2012.120
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2012.120
Keywords
This article is cited by
-
Pharmacokinetics, mass balance, and metabolism of [14C]vicagrel, a novel irreversible P2Y12 inhibitor in humans
Acta Pharmacologica Sinica (2021)
-
Hormonal, chemical and thermal inhibition of spermatogenesis: contribution of French teams to international data with the aim of developing male contraception in France
Basic and Clinical Andrology (2017)
-
World Heart Federation expert consensus statement on antiplatelet therapy in East Asian patients with ACS or undergoing PCI
Nature Reviews Cardiology (2014)
-
Pharmacometrics: a quantitative tool of pharmacological research
Acta Pharmacologica Sinica (2012)
