
Abstract
Aim:
To investigate the anti-arrhythmic effects of sulfamide analogues of changrolin and to characterize the sulfate of compound 6f (sulcardine sulfate, Sul) as a novel anti-arrhythmic agent.
Methods:
The anti-arrhythmic effects of compounds were studied against aconitine-induced arrhythmias in rats and ouabain-induced arrhythmias in guinea pigs. The effects of Sul on transmembrane action potentials were investigated in isolated rabbit sinoatrial nodes and guinea-pig papillary muscles using intracellular recording. With a whole-cell recording technique, the effects of Sul on sodium current, calcium current, and potassium currents were examined in isolated single guinea-pig ventricular myocytes.
Results:
In aconitine-induced arrhythmias of rats, sulfamide analogues of changrolin (4, 5, and 6a–6p) exhibited various anti-arrhythmic activities. The sulfate of compound 6f (Sul) increased the amount of aconitine required to induce arrhythmias in each treated animal. The ED50 value of Sul in rats was 196 mg/kg. In ouabain-induced arrhythmias of guinea pigs, 25, 50, and 100 mg/kg doses of Sul increased the dose of ouabain required to induce VP, VT, and VF in a dose-dependent manner. In papillary preparations, Sul produced a concentration-dependent decrease in APA and Vmax, prolonged APD90 and ERP, whereas RP was unaffected. In the spontaneously beating sinus nodes, Sul reduced APA and Vmax in a concentration-dependent manner. The whole-cell recording studies revealed that Sul produced a reversible reduction in INa (IC50=26.9 μmol/L) and ICa,L(IC50=69.2 μmol/L), whereas the inward rectifier (IK1) and the delayed rectifier potassium currents (IK) were unaffected.
Conclusion:
As a multi-ion channel blocker, Sul may have potent efficacy in anti-atrial and ventricular arrhythmias.
Similar content being viewed by others

Introduction
The use of anti-arrhythmic drugs has decreased over the last 20 years because of the side effect of pro-arrhythmia, more serious rhythm disorders induced by some anti-arrhythmic agents. The majority of anti-arrhythmic agents are ion channel blockers. The Cardiac Arrhythmia Suppression Trial (CAST) and subsequent CAST II studies have provided evidence indicating that these drugs primarily act by blocking sodium ion channels and may have the potential to increase mortality in patients with structural heart disease1. Since then, the study of anti-arrhythmic agents has shifted away from sodium ion channel blockers and focused on potassium channel blockers, such as amiodarone and sotalol derivatives, which have been proven to be highly effective agents for treating life-threatening arrhythmias. However, no selective channel blocker has yet been shown to unequivocally reduce the mortality associated with atrial or ventricular arrhythmias. Recent studies have shown that amiodarone, an agent with a broad range of effects, including the blocking of sodium, calcium and delayed rectifier potassium current (Ikr) channels and non-competitive β-blockade, can prolong the QT interval but rarely causes pro-arrhythmia. The likely explanation for this paradox is that mixed or multi-ion channel blocking effects decrease the pro-arrhythmia side effect2. However, amiodarone therapy has other serious side effects such as pulmonary, skin and thyroid toxicities. Therefore, there is a major need for new drugs that will control arrhythmias more safely and effectively. Based on clinical data, compounds with appropriate mixed channel blocking effects may be a promising route to new anti-arrhythmic drugs3.
Since the 1980s, implantable cardioventer-defibrillators have been an option for treating arrhythmia. However, an adjuvant drug therapy is often required to provide maximum prevention and treatment of life-threatening arrhythmias.
It is well-known that the anti-malarial drug quinine and its optical isomer quinidine have anti-arrhythmic properties. The demonstration of both anti-malarial and anti-arrhythmic activities is not confined to quinine and quinidine. Febrifugine is an alkaloid isolated from Dichroa febrifuga that was used against malaria in traditional Chinese medicine. In the 1970s, changrolin (1), a compound derived from febrifugine, was found to have anti-arrhythmic properties in malaria patients with arrhythmia and markedly diminished ectopic beats in patients. In subsequent investigations of its anti-arrhythmic effects, changrolin exhibited significant protective and therapeutic effects against arrhythmia, both in animal models and in clinical trials. In animal studies, the anti-arrhythmic potency of changrolin was higher and its toxicity was lower than those of quinidine. Changrolin effectively prevented atrial fibrillation induced by acetylcholine in rats and elevated the threshold of electrically induced ventricular fibrillation in rabbits and dogs. The results of clinical trials on 489 patients with various arrhythmias demonstrated that changrolin was most effective in reducing paroxysmal ventricular tachycardia and ventricular premature beats by 85.7% and 84.3%, respectively4, 5. However, changrolin also exhibited some side effects, including skin pigmentation and parasympatholytic activity4. Aqueous solutions of changrolin are unstable and turns brownish-yellow after storage at room temperature for one or two days. The chemical structure of changrolin is unlike those of currently marketed anti-arrhythmic drugs, and a number of analogues and derivatives of changrolin have been prepared and tested for anti-arrhythmic activity6, 7, 8, 9, 10, 11, 12, 13.
Structurally, changrolin could be divided into three portions: a quinazoline moiety, a 3,5-bis(1-pyrrolidylmethyl)-4-hydroxy phenyl moiety and a linker between the two moieties. In 1981, Sun et al reported that the benzoyl analogue of changrolin, compound 2, exhibited protective effects against experimental arrhythmia induced by aconitine. Compound 2 was more potent than changrolin in protecting dogs from atrial fibrillation induced by acetylcholine6, 7. Subsequently, Stout et al reported an amide analogue 3, which is the amide-reversed form of amide 2. Compound 3 possessed excellent profiles as a class 1 anti-arrhythmic11. All of the SAR data reported so far regarding the structure-activity relationships of changrolin can be summed as follows:
-
1)
The 3,5-bis(1-pyrrolidylmethyl)-4-hydroxyphenyl moiety is essential for optimal anti-arrhythmic activity; pyrrolidine yields greater activity than other secondary amines.
-
2)
The quinazoline moiety could be replaced by a variety of aromatic or heteroaromatic rings without loss of activity. The position and type of substituents on the rings did not affect activity.
-
3)
The −NH-linker could be replaced with −CH2O−, −NHCO−, −CONH–, −CONHCH2–, and −CO−. Some amides were found to be potent anti-arrhythmic agents.
-
4)
The quinazoline moiety and −NH-linker could be simultaneously replaced by −(CH2)nCOOR group without loss of activity (Figure 1).
Figure 1 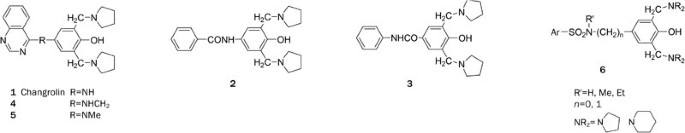
Chemical structures of compound 1–6.

Based on the reported structure-activity relationship data for changrolin, further structural modifications were performed by our groups. The purpose of this study was to assess the protective and therapeutic effects against arrhythmia of the new analogues of changrolin14.
Materials and methods
Chemistry
The synthesis of all target compounds (4, 5, 6a–6p) is straightforward. The preparation of compounds 4 and 5 is depicted in Scheme 1. Chloroquinazoline was coupled with 4-hydroxybenzylamine or N-methyl-4-hydroxyaniline to give compound 7 or 8. The target compounds 4 and 5 were obtained by Mannich reaction of 7 and 8, respectively, with formaldehyde and pyrrolidine.

Synthesis of compounds 4 and 5.
Sulfamides 6a, 6e, 6h, and 6k were prepared by the reaction of 4-amino-2,6-bis(1-pyrrolidylmethyl)phenol (9)9 with the corresponding 4-substituted benzenesulfonyl chloride (Scheme 2).

Preparation of sulfamides 6a, 6e, 6h, and 6k.
Sulfamides 6b, 6f, 6i, and 6m, the methylene insertion analogues of compounds 6a, 6e, 6h, and 6k, were obtained by sulfonylation of 4-hydroxybenzylamine with 4-substituted benzenesulfonyl chlorides in the presence of triethylamine, followed by Mannich reaction of the resulting sulfamides 10–13 with formaldehyde and pyrrolidine in ethanol (Scheme 3).

Synthesis of the methylene insertion sulfamides 6b, 6f, 6i, and 6m.
Using N-methyl-4-hydroxy-aniline instead of 4-hydroxybenzylamine, the N-methyl analogues 6c, 6g, 6j, and 6n were synthesized in the same manner as described in Scheme 4.

Synthesis of N-methyl analogues 6c, 6g, 6j, and 6n.
The N-ethyl analogue 6d was prepared by coupling 4-toluenensulfonyl chloride with 4-ethylamino-2,6-bis(1-pyrrolidylmethyl)phenol (18) in the presence of triethylamine in ethanol (Scheme 5).

Synthesis of N-ethylsulfamide 6d.
The two 1-naphthalenesulfamides 6o and 6p were yielded via Mannich reaction of N-(4-hydroxyphenyl)-1-naphthalenesulfamide with formaldehyde and pyrrolidine or peperidine, respectively.
Drugs and reagents
Sulfamide analogues of changrolin and sulcardine sulfate (6f, a yellowish powder with purity over 98.5%, MW=611.76) were synthesized by the chemists in our groups. Aconitine, ouabain, and collagenase (type I) were purchased from Sigma Chemical Co (St Louis, MO, USA). Other reagents, unless otherwise specified, were commercial products of reagent grade and purchased from Sigma-Aldrich China, Inc (Shanghai, China).
Animals
Mice, Sprague Dawley rats, and guinea pigs were obtained from the Shanghai Laboratory Animal Center (Shanghai, China). The animals were cared for in accordance with the institutional guidelines of the Animal Care and Use Committee of the Shanghai Institute of Materia Medica, Chinese Academy of Sciences.
Aconitine-induced arrhythmias in rats
Sprague Dawley rats of either sex weighing 220–250 g were used. The animals were anesthetized by intraperitoneal injection of 1.2 g/kg urethane. Aconitine (10 μg/mL) was administered through continuous infusion into the saphenous vein at a rate of 0.08 mL/min, and the electrocardiogram (ECG) in lead II was recorded. The test compound was given orally 2 h prior to aconitine infusion. The time until the onset of the first ventricular premature (VP), ventricular tachycardia (VT), and ventricular fibrillation (VF) was chosen as the endpoint, and the amount of aconitine required to induce the arrhythmias was calculated. The percent increase in the amount of aconitine required to induce arrhythmias in animals treated with each test compound (above the mean value in the appropriate control group) was calculated, and log dose-response curves were constructed by liner regression analysis. ED50 values were defined as the dose of test compound required to increase the arrhythmogenic dose of aconitine by 50% relative to that required in control animals.
Ouabain-induced arrhythmias in guinea pigs
Guinea pigs of either sex weighing 250–300 g were anesthetized with 1.2 g/kg urethane intraperitoneally. One jugular vein was catheterized. Ouabain (300 μg/mL) was infused into the jugular vein at a rate of 3 μg/min. The ECG in lead II was recorded with subcutaneous steel-needle electrodes. The time until the onset of the first VP, VT, and VF was chosen as the endpoint, and the amount of ouabain required to induce the arrhythmias was calculated.
Prophylactic index and therapeutic index
The prophylactic index and therapeutic index were obtained by the following procedures: the performance of an acute toxicity test in mice to obtain the medium lethal dose (LD50); the use of an aconitine-induced arrhythmia model in rats to obtain the medium effective dose (ED50) in prophylactic tests and the effective dose (ED) in therapeutic tests; and the calculation of the prophylactic indexes as the ratio LD50:ED50 and the therapeutic index as the ratio LD50:ED. Both indexes were used to evaluate the compounds for their anti-arrhythmic activities15, 16.
Action potential recording in isolated guinea-pig papillary muscles
The guinea pig papillary muscle was dissected from the right ventricle and superfused in a 1.5 mL chamber (15 mL/min) with modified Tyrode's solution gassed with 95% O2/5% CO2 at 32±0.5 °C17. The composition of the modified Tyrode's solution was as follows: 120.3 mmol/L NaCl, 4.0 mmol/L KCl, 1.2 mmol/L CaCl2, 1.3 mmol/L MgSO4, 25.2 mmol/L NaHCO3 and 5.5 mmol/L glucose (pH 7.4). The preparations were allowed to equilibrate for 1 h before recording the control. Action potentials (APs) were recorded using glass microelectrodes filled with 3 mol/L KCl and a tip resistance of 10–20 MΩ. Signals were delivered to a microelectrode amplifier (MEZ-7101, Nihon Kohden, Japan). The maximum upstroke velocity of AP (Vmax) was measured with a PowerLab System (AD Instruments Pty Ltd, Australia). Both AP and Vmax were monitored on the PowerLab System. The APs were elicited with rectangular pulses (1 ms, twice the threshold) generated by a stimulator (SEN-7203, Nihon Kohden, Japan) at 1 Hz.
Isolation of ventricular myocytes
The enzymatic dissociation method was described previously18, 19. In brief, the excised guinea pig heart was perfused through the aorta retrogradely with a Ca2+-free Tyrode's solution for 10 min at 37 °C, followed by a Tyrode's solution containing 0.2 mg/mL collagenase (type I) for 3–4 min. The composition of the Tyrode's solution was as follows: 135 mmol/L NaCl, 5.4 mmol/L KCl, 1 mmol/L MgCl2, 0.33 mmol/L NaH2PO4, 5 mmol/L HEPES and 5 mmol/L glucose (pH 7.4); the solution was oxygenated with 100% O2. The ventricles were then minced and gently triturated for 10 min in Kraft-Brühe (KB) solution containing the following: 50 mmol/L L-glutamic acid, 80 mmol/L KOH, 40 mmol/L KCl, 3 mmol/L MgSO4, 25 mmol/L KH2PO4, 10 mmol/L HEPES, 1 mmol/L EGTA, 20 mmol/L taurine, and 10 mmol/L glucose (pH 7.4). Myocytes were harvested with 200 μm nylon mesh and stored in KB solution at 20–24 °C.
Voltage-clamp recording
A cell suspension was placed in a 3 mL chamber mounted on the stage of a microscope (Optiphot-2; Nikon, Japan) and superfused with an external solution via a PBS-8 solution exchange system (ALA Scientific Instruments Inc, USA) at a rate of 3 mL/min. Patch pipettes were pulled with a P-97 microelectrode puller (Shutter Instruments Co, USA), and had a tip resistance of 1–5 MΩ when filled with a pipette solution. Whole-cell voltage-clamp recording was made from single ventricular myocytes with an Axopatch-1D amplifier (Axon Instruments, USA). After a gigaseal formation (seal resistance 1 GΩ), the membrane was ruptured with gentle suction to obtain the whole-cell voltage-clamp configuration. Voltage command protocols were provided by the pClamp 6.0.4 software package (Axon Instruments, USA) via a DigiData-1200 interface (Axon Instruments, USA). Capacitance compensation was routinely optimized, and series resistance was compensated by 40%–80%. Linear leaks were subtracted digitally online. Signals were filtered at 3 kHz, digitized at 10 kHz, and stored in an IBM-compatible computer. Cell capacitance was measured with a short hyperpolarizing ramp pulse (5 mV in 5 ms) from a holding potential of -40 mV. The membrane capacitance of myocytes ranged from 100–150 pF. The Na+ current (INa), L-type Ca2+ current (ICa,L) and inward rectifier K+ current (IK1) were recorded at 20–24 °C, whereas the delayed rectifier K+ current (IK) was recorded at 35±1 °C.
The composition of the external solution for recording INa was as follows: 120 mmol/L tetraethylammonium chloride (TEA-Cl), 30 mmol/L NaCl, 1 mmol/L MgCl2, 1 mmol/L CaCl2, 0.1 mmol/L CdCl2, 10 mmol/L HEPES, and 10 mmol/L glucose (pH 7.4). The external solution used to record ICa,L contained the following: 120 mmol/L TEA-Cl, 1 mmol/L MgCl2, 10 mmol/L CsCl, 1.8 mmol/L CaCl2, 10 mmol/L HEPES and 10 mmol/L glucose (pH 7.4). The external solution used to record the K+ currents contained the following: 140 mmol/L NaCl, 5.4 mmol/L KCl, 1 mmol/L MgCl2, 0.1 mmol/L CaCl2, 0.1 mmol/L CdCl2, 5 mmol/L HEPES, and 5 mmol/L glucose (pH 7.4). The pipette solution for recording INa contained the following: 80 mmol/L CsCl, 40 mmol/L CsOH, 10 mmol/L EGTA, 10 mmol/L HEPES, and 5 mmol/L Na2ATP (pH 7.2). The pipette solution for recording ICa,L contained the following: 80 mmol/L CsCl, 40 mmol/L CsOH, 10 mmol/L EGTA, 10 mmol/L HEPES, and 5 mmol/L MgATP (pH 7.2), while that for recording the K+ currents contained the following: 140 mmol/L KCl, 0.5 mmol/L MgCl2, 10 mmol/L EGTA, 10 mmol/L HEPES, and 5 mmol/L K2ATP (pH 7.2). The myocytes were perfused with external solutions and allowed to equilibrate for about 5 min with ascending concentrations of drug to generate concentration-response relationships.
Statistical analysis
Data are presented as the mean±SD. A two-tailed Student's t-test was used for analyses of anti-arrhythmic activity and AP measurements. The patch-clamp data were analyzed using the Clampfit 9.0 (Axon Instruments, USA) and Prism 3.0 software (GraphPad Software, USA). For all tests, P<0.05 was considered statistically significant. The concentration of sulcardine sulfate that yielded a 50% inhibitory effect (IC50 value) was obtained by fitting the concentration–response relationship to the equation: X=1/{1+([C]/IC50)N}, where X is the normalized response, [C] is the concentration of the agent, and N is the Hill coefficient.
Results and discussion
Anti-arrhythmic activity of sulfamide analogues (6a–6p) of changrolin (1)
It was supposed that the aminophenol moiety in changrolin could be oxidized into an iminoquinone unit, resulting in an extended conjugated system with quinazoline in vivo, which may be responsible for both the skin pigmentation in patients after long-term administration of changrolin and the darkening of the aqueous solution of changrolin at room temperature in vitro. Because both bis(pyrrolidylmethyl)phenol and heteroaromatic or aromatic moieties in the changrolin molecule are essential for anti-arrhythmic activity, two approaches could be adopted to avoid the formation of the extended iminoquinone-containing conjugate chromophore. First, the −NH-linkage of the two aromatic rings in changrolin could be replaced by −NHCH2− or −NMe-, giving compounds 4 and 5, respectively. An alternative approach was the replacement of the quinazolylamino moiety in changrolin with an arylamido group, such as in compounds 2 and 3, both of which have been reported to show potent anti-arrhythmic activities6, 7, 11. Considering that sulfamide is the isostere of amide, we focused on the analogues with a −SO2NH-linkage instead of −CONH− in compound 2 and with an additional −CH2− unit between the sulfamido and bis(1-pyrrolidylmethyl)phenol moiety (Table 1). A new series of aromatic sulfamide analogues of changrolin, 6a–6p, was thus designed and prepared.
Two analogues, 4 and 5, derived from changrolin by inserting a methylene between the amino group and the phenol ring and by methylation of the amino group in changrolin, respectively, were first designed and synthesized. The anti-arrhythmic activities of 4 and 5 were less potent than that of changrolin itself. The pharmacological screening data of sulfamides 6a–6p showed that most of the sulfamides possessed anti-arrhythmic activity (Table 1). Among them, compound 6f (sulcardine) was the best one. Compounds with one carbon extension (6i and 6m) or N-methyl substitution (6j and 6n) did not change the potency markedly relative to 6h and 6k, respectively. When the 1-pyrrolidyl group in 6o was replaced by 1-piperidyl (6p), the anti-arrhythmic activity increased. In the 4-methoxybenzenesulfamides group (6e, 6f, 6g), the insertion of a methylene unit between the sulfamido and phenol ring enhanced the prophylactic index up to 6-fold (6f vs 6e). N-Methylation of 6e increased the prophylactic index by 2.3-fold (6g vs 6e). In the 4-methylbenzensulfamides group, although all of the compounds tested (6a–6d) exhibited anti-arrhythmic activity, the results were ambiguous. Inserting a methylene unit between the sulfamido and phenol ring in 6a resulted in a slight change in both the prophylactic index and the therapeutic index (6a vs 6b). N-Methylation of 6a caused a 3.7-fold increase in the prophylactic index and a 5.5-fold decrease in the therapeutic index (6c vs 6a). However, N-ethylation of 6a resulted in a 5-fold decrease in both the prophylactic index and the therapeutic index (6a vs 6d).
The prophylactic indexes of the naphthylenesulfamides (6o and 6p) were much higher than those of the benzenesulfamides, with the exception of 6f (sulcardine). There was no obvious correlation of anti-arrhythmic activity with electron-donating or electron-withdrawing substituents on the benzene ring (6a, 6e, and 6k vs 6h; 6b, 6m vs 6i; 6c, 6g, and 6n vs 6j). Surprisingly, 6f (sulcardine) had a much higher prophylactic index than 6b. The optimal spectrum of prophylactic index and therapeutic index was given by 6f (sulcardine).
To summarize our findings on the chemical modifications of the structure of changrolin (1), we have found that the quinazoline moiety could be replaced by an aromatic sulfonyl group without loss of activity. The insertion of a methylene unit between the sulfamido and phenol ring and N-methylation of sulfamido resulted in the retention of or an increase in anti-arrhythmic activity.
Effects of compound 6f sulfate (sulcardine sulfate) on experimental arrhythmia models in animals
Compound 6f sulfate was tested in various arrhythmia models in animals. Against aconitine-induced arrhythmias in rats, 6f sulfate (100, 150, and 250 μg/kg) significantly increased the amount of aconitine required to induce arrhythmias at each dose tested (Table 2). The ED50 value of 6f sulfate in rats was 196 mg/kg. At 2, 3, 4, and 6 h after administration, 6f sulfate (at a dose of 1.5-fold ED50) significantly raised the arrhythmogenic dose of aconitine to induce VP, VT, and VF in rats. The effects of 6f sulfate reached a peak at 2–3 h and lasted up to 6 h after administration (Table 3).
Against ouabain-induced arrhythmias in guinea pigs, 6f sulfate at 25, 50, and 100 mg/kg increased the dose of ouabain required to induce VP from 134±8 μg/kg to 156±9, 176±16, and 187±14 μg/kg, respectively. The amount of ouabain required to induce VT and VF was also increased as the administered dose of 6f sulfate increased (Table 4).
Effects of compound 6f sulfate (sulcardine sulfate) on action potentials
Guinea pig papillary muscle cells are characterized by firing fast response action potentials. The application of 6f sulfate (10, 30, and 100 μmol/L) reduced the action potential amplitude (APA) and the maximum upstroke velocity of the action potential (Vmax) and prolonged the action potential duration at 90% repolarization (APD90) as well as the effective refractory period (ERP) (Table 5). In rabbit sinoatrial nodal dominant pacemaker cells, which are characterized by firing slow response action potentials, the application of 6f sulfate (10, 30, and 100 μmol/L) reduced Vmax and the slope of phase 4 depolarization and decreased APA. At 100 μmol/L, 6f sulfate also prolonged APD, decreased MRP and SEF. The changes were 5.5%, 11.6%, and 4.7%, respectively (n=5, P<0.05) (Table 6).
Inhibition of sodium current and L-type calcium current in ventricular myocytes
The above results suggest that 6f sulfate is a mixed or multi-ion channel blocker against Na+, Ca2+, and K+ channels20. Thus, the actions of 6f sulfate on cardiac ion channels were studied with the whole-cell patch clamp method in isolated ventricular myocytes. The application of 6f sulfate caused a reversible reduction in INa (IC50=26.9 μmol/L) and ICa,L(IC50=69.2 μmol/L), whereas the inward rectifier (IK1) and the delayed rectifier potassium currents (IK) were unaffected. The inhibition of voltage-gated sodium and calcium channels induced by 6f sulfate may contribute to its effect on atrial and ventricular tachyarrhythmias.
An overall preclinical examination of 6f sulfate demonstrated that this compound possesses a promising profile as an anti-arrhythmic agent. Thus, the phase I clinical trial of this compound is complete, and the data indicate that this compound may be a novel anti-arrhythmic drug with low pro-arrhythmic side effects.
Conclusion
We have designed and synthesized a series of sulfamide analogues of changrolin (1) as potential anti-arrhythmic agents. To avoid the formation of the extended conjugation system from the changrolin molecule in vivo, which may be the cause of skin pigmentation, changrolin was chemically modified by two routes. One was the replacement of the quinazoline moiety by aromatic sulfonyl groups; the other was the insertion of a methylene unit between the NH group and phenol ring or N-alkylation of the NH group.
Pharmacological screenings of the target compounds (4, 5, and 6a–6p) demonstrated that all of the target compounds possessed various anti-arrhythmic activities. A considerable improvement of the anti-arrhythmic potency of these compounds compared to changrolin was observed. Based on both the prophylactic and therapeutic indexes, the target compounds with better anti-arrhythmic activity are 6a, 6f, 6i, 6o, and 6p. Among these compounds, compound 6f is exceptionally potent and was chosen for considera tion as a drug candidate. The preclinical and phase I clinical studies of 6f in the sulfate trihydrate form are complete. The compound has been named sulcardine sulfate and is well-tolerated in phase I trials. Sulcardine sulfate is now undergoing phase II trials. The chemical structure of 6f is different from marketed anti-arrhythmic drugs. As a multi-ion channel blocker, it may have potent efficacy in anti-atrial fibrillation and anti-ventricular tachycardia, which are two particularly important cardiac arrhythmias with important clinical implications.
Experimental section
General methods
The 1H NMR spectra were recorded on Bruker Am-400 and Bruker Ac-100 NMR spectrometers. The data are reported in parts per million relative to TMS and are referenced to the solvent in which they were analyzed. Elemental analyses were obtained on an Elemental Vario EL instrument. Melting points were determined on a Buchi-510 capillary apparatus and are uncorrected. IR spectra were recorded on a Perkin-Elmer-599B spectrometer. The solvent was removed by rotary evaporation under reduced pressure, and flash column chromatography was performed on silica gel (200–300 mesh) from the Qingdao Haiyang Chemical Company. Anhydrous solvents were obtained by redistillation over sodium wire.
4-[3,5-bis(1-pyrrolidylmethyl)-4-hydroxybenzyl]aminoquinazoline (4)
A mixture of 4-chloroquinazoline (1.51 g, 9.12 mmol) and 4-hydroxybenzylamine (2.82 g, 22.9 mmol) in ethanol (6 mL) was refluxed for 3 h. After cooling, the precipitate was collected and washed successively with a saturated solution of NaHCO3 and water. The solid was recrystallized from EtOH-H2O to give 4-(4-hydroxybenzyl)aminoquinazoline 7 (1.30 g, 56%), mp 231–232 °C. 1H NMR (CDCl3) δ 4.58 (s, 2H), 6.58 (d, J=8.4 Hz, 2H), 7.33 (d, J=8.4 Hz, 2H), 7.35 (m, 1H), 7.53 (d, J=8.7 Hz, 1H), 7.61 (m, 1H), 7.97 (d, J=8.8 Hz, 1H), 8.27 (s, 1H). Anal. (C15H13N3O·H2O) C, H, N.
A mixture of 7 (1.30 g, 5.2 mmol), pyrrolidine (1.1 mL, 13.2 mmol) and a 36%–38% solution of formaldehyde (1.1 mL, 15 mmol) in ethanol (10 mL) was refluxed for 3 h. The solvent was removed under reduced pressure to give an oil that became crystalline upon standing. Recrystallization from EtOH-H2O yielded 4 (1.35 g, 63%), mp 155–156 °C . 1H NMR (CDCl3) δ 1.60–1.90 (m, 8H), 2.50–2.80 (m, 8H), 3.72 (s, 4H), 4.75 (s, 2H), 7.00–7.80 (m, 6H), 8.60 (s, 1H). Anal. (C25H31N5O) C, H, N.
4-N-[3,5-bis(1-pyrrolidylmethyl)-4-hydroxyphenyl]-N-methylaminoquinazoline (5)
In the same manner as in the preparation of 7 and 4, 4-N-methyl-(4-hydroxyphenyl)aminoquinazoline (8) was obtained from 4-chloroquinazoline and N-methylaminophenol in 65% yield, mp 258–260 °C (DMF-EtOH). 1H NMR (CDCl3) δ 3.25 (s, 3H), 7.03 (d, J=8.5 Hz, 2H), 7.40 (d, J=8.5 Hz, 2H), 8.00–8.60 (m, 4H), 8.78 (s, 1H). Anal. (C15H13N3O) C, H, N.
Mannich reaction of 8 gave 5 in 51% yield. The crude product was first purified by column chromatography (silica gel, EtOH:25% NH4OH=10:0.25), and recrystallized from EtOH-H2O, mp 129–130 °C. 1H NMR (CDCl3) δ 1.55–1.80 (m, 8H), 2.30–2.60 (m, 8H), 3.45 (s, 3H), 3.62 (s, 4H), 6.90–7.80 (m, 6H), 8.50 (s, 1H). Anal. (C25H31N5O) C, H, N.
N-[3,5-bis(1-pyrrolidylmethyl)-4-hydroxyphenyl]-4-chlorobenzenesulfamide (6h)
To a solution of 2,6-bis-(1-pyrrolidiylmethyl)-4-aminophenol (1.3 g, 4.8 mmol) in tetrahydrofuran (5 mL) was added dropwise a solution of 4-chlorobenzenesulfonyl chloride (1.0 g, 4.8 mmol) in tetrahydrofuran (5 mL). The mixture was stirred at room temperature for 4 h. The solvent was removed in vacuo, and the residue was dissolved in water. After neutralization with 2 mol/L KOH solution, the mixture was extracted with ethyl acetate. The organic layer was successively washed with water and brine and dried over anhydrous Na2SO4. After removing the solvent in vacuo, the residue was purified by column chromatography (silica gel, CH3COOEt:CH3OH:25% NH4OH=9:1:0.05). The resultant solid was recrystallized from ethyl acetate to give the product 6h (1.3 g, 60%), mp 187–188 °C. 1H NMR (CDCl3) δ 1.72–2.08 (m, 8H), 2.56–2.88 (m, 8H), 3.88 (s, 4H), 6.90 (s, 2H), 7.36 (d, J=8.7 Hz, 2H), 7.64 (d, J=8.7 Hz, 2H). Anal. (C22H28N3ClO3S) C, H, N, Cl.
Compounds 6a, 6e, and 6k were prepared in the same manner as 6h.
N-[3,5-bis(1-pyrrolidylmethyl)-4-hydroxyphenyl]-4-methylbenzenesulfamide (6a)
Yield: 47%, mp 160–161 °C (EtOH-H2O). 1H NMR (CDCl3) δ 1.76–2.12 (m, 8H), 2.38 (s, 3H), 2.72-3.08 (m, 8H), 3.96 (s, 4H), 6.99 (s, 2H), 7.18 (d, J=8.6 Hz, 2H), 7.56 (d, J=8.6 Hz, 2H). Anal. (C23H31N3O3S) C, H, N.
N-[3,5-bis(1-pyrrolidylmethyl)-4-hydroxyphenyl]-4-methoxybenzenesulfamide (6e)
Yield: 79%, white crystals, mp 143–144 °C (CH3COOEt). 1H NMR (CDCl3) δ 1.64–1.94 (m, 8H), 2.35–2.65 (m, 8H), 3.65 (s, 3H), 3.81 (s, 4H), 6.75 (s, 2H), 6.85 (d, J=8.7 Hz, 2H), 7.58 (d, J=8.7 Hz, 2H). Anal. (C23H31N3O4S) C, H, N.
N-[3,5-bis(1-pyrrolidylmethyl)-4-hydroxyphenyl]-4-acetamidobenzenesulfamide (6k)
Yield: 85%, mp 186–187 °C (CH3COOEt). 1H NMR (CDCl3) δ 1.82–2.06 (m, 8H), 2.17 (s, 3H), 2.83–3.07 (m, 8H), 3.76 (s, 4H), 7.09 (s, 2H), 7.54 (d, J=8.6 Hz, 2H), 7.65 (d, J=8.7 Hz, 2H), 9.50 (s, 1H). Anal. (C24H32N4O4S) C, H, N.
N-[3,5-bis(1-pyrrolidylmethyl)-4-hydroxybenzyl]-4-methoxybenzenesulfamide (sulcardine, 6f) and the sulfate (sulcardine sulfate)
-
1)
To a suspension of 4-hydroxybenzylamine (133 g, 1.08 mol) in DMF (500 mL) was added dropwise 4-methoxybenzensulfonyl chloride (206 g, 1.00 mol) in DMF (320 mL) over a period of 30 min at 0–10 °C with stirring, followed by the addition of triethylamine (158 mL, 1.12 mol) over 30 min at the same temperature. The stirring was continued for an additional 1.5 h at room temperature. The reaction mixture was poured into ice-water (5 L). After stirring for 10 min, the suspension was allowed to stand for 2 h. The solid was filtered, washed with water (300 mL×3), and dried in a desiccator over anhydrous calcium chloride, yielding N-(4-hydroxybenzyl)-4-methoxybenzenesulfamide (11) (248 g, 85%) as a white solid, mp 160–162 °C. The authentic sample was obtained by recrystallization from ethyl acetate, mp 161–162 °C. 1H NMR (CD3OD) δ 3.70 (s, 3H), 3.76 (s, 2H), 6.48 (d, J=8.4 Hz, 2H), 6.82(d, J=8.4 Hz, 2H), 6.86 (d, J=8.7 Hz, 2H), 7.56 (d, J=8.7 Hz, 2H). EIMS (m/z): 293 (M+), 254, 195, 185, 171, 155, 149, 122 (100), 107, 99, 77, 65. Anal. (C14H15NO4S) C, H, N.
-
2)
A mixture of 11 (230 g, 0.78 mmol), pyrrolidine (200 mL, 2.44 mol) and 36% aqueous formaldehyde (250 mL, 3.30 mol) in ethanol (800 mL) was stirred under reflux for 8 h. The reaction mixture was concentrated under vacuum to dryness. The resulting oil residue was dissolved in chloroform (350 mL), and the solution was washed with water (300 mL×3). Under stirring, the organic layer was mixed with water (300 mL), and then concentrated hydrochloric acid (approximately 165 mL) was added portionwise at 0-10 °C to adjust the pH of the aqueous phase to ∼2. The aqueous phase was washed with chloroform (200 mL) and then mixed with additional chloroform (300 mL). Under stirring, the two-phase mixture was treated portionwise with 25%–28% aqueous ammonia (∼300 mL) to adjust the pH of the aqueous phase to 9–10. The organic layer was separated, and the aqueous layer was further extracted with chloroform (200 mL×2). The combined organic layer was dried over anhydrous sodium sulfate and concentrated under vacuum to dryness. The oily residue was treated with acetone (45 mL) and isopropyl ether (290 mL), and the mixture was heated under reflux until the suspension became a solution. The solution was cooled to room temperature, seeded with an authentic sample, and allowed to stand at 0 °C overnight. The solid was filtered and dried under vacuum, yielding product 6f (290 g, 81%) as a yellowish solid, mp 96–98 °C. The authentic sample was obtained by preparative TLC or column chromatography (silica gel; CHCl3:MeOH:25% NH4OH=92:7:1). The compound could be recrystallized from ethanol-water, mp 101–102 °C. 1H NMR (CDCl3) δ 1.77–1.86 (m, 8H), 2.53–2.63 (m, 8H), 3.68 (s, 4H), 3.86 (s, 3H), 3.97 (s, 2H), 6.86 (s, 2H), 6.95 (d, J=8.7 Hz, 2H), 7.78 (d, J=8.6 Hz 2H). EIMS (m/z): 459 (M+), 390, 388, 202, 171, 148, 107, 84, 70 (100). Anal. (C24H33N3O4S) C, H, N.
-
3)
Under stirring, the Mannich base 6f (150.5 g, 0.327 mol) was mixed with 2 mol/L H2SO4 (172 mL, 0.344 mol), and the mixture was heated at 80 °C until the solid dissolved. The solution was cooled to room temperature, seeded with an authentic sample, and the sulfate of 6f was formed as crystals. To the stirred mixture was added anhydrous ethanol (520 mL), and the mixture was allowed to stand at 0 °C for 24 h. The solid was filtered, washed with ethanol, and recrystallized with 80% ethanol (250 mL). The sulfate was dried over concentrated sulfuric acid in a desiccator, giving the sulfate of 6f (143 g, 71%) as a trihydrate, mp 125–140 °C. 1H NMR (D2O) δ 2.00–2.13 (m, 4H), 2.14–2.25 (m, 4H), 3.12–3.22 (m, 4H), 3.45–3.55 (m, 4H), 3.90 (s, 3H), 4.20 (s, 2H), 4.33 (s, 4H), 7.06 (d, J=8.7 Hz, 2H), 7.28 (s, 2H), 7.66 (d, J=8.9 Hz, 2H). 13C NMR (D2O) δ 24.7, 47.6, 55.7, 56.1, 58.1, 116.6, 122.5, 131.3, 132.3, 133.3, 136.0, 155.8, 164.8. EIMS (m/z): 459, 390, 388, 202, 171, 148, 107, 84, 70 (100). Anal. (C24H33N3O4S·H2SO4·3H2O) C, H, N, S.
Compounds 10, 12, 13, 6b, 6i, and 6m were also prepared by the above-mentioned procedures for 11 and 6f.
N-[3,5-bis(1-pyrrolidylmethyl)-4-hydroxybenzyl]-4-methylbenzenesulfamide (6b)
N-(4-hydroxybenzyl)-4-methylbenzenesulfamide (10) was obtained in 73% yield, mp 183–184 °C (EtOH). 1H NMR (CDCl3) δ 2.45 (s, 3H), 3.96 (s, 2H), 6.70 (d, J=8.6 Hz, 2H), 7.03 (d, J=8.5 Hz, 2H), 7.31 (d, J=8.7 Hz, 2H), 7.72 (d, J=8.8 Hz, 2H). Anal. (C14H15NO3S) C, H, N.
Mannich reaction of 10 afforded the crude product 6b. It was purified by column chromatography (silica gel, AcOEt:MeOH:25% NH4OH=1:1:0.1) and recrystallized from EtOH-H2O to give 6b in 80% yield, mp 129–130 °C. 1H NMR (CDCl3) δ 1.68–1.88 (m, 8H), 2.41 (s, 3H), 2.44–2.64 (m, 8H), 3.63 (s, 4H), 3.70 (s, 2H), 6.84 (s, 2H), 7.28 (d, J=8.7 Hz, 2H), 7.72 (d, J=8.8 Hz, 2H). Anal. (C24H33N3O3S) C, H, N.
N-[3,5-bis(1-pyrrolidylmethyl)-4-hydroxybenzyl]-4-chlorobenzenesulfamide (6i)
N-(4-hydroxybenzyl)-4-chlorobenzenesulfamide (12) was obtained in 73% yield, mp 187–189 °C (EtOH). 1H NMR (CDCl3) δ 3.14 (s, 2H), 6.76 (d, J=8.7 Hz, 2H), 6.95 (d, J=8.7 Hz, 2H), 7.51 (m, 4H). Anal. (C13H12NO3SCl) C, H, N, Cl.
Mannich reaction of 12 gave product 6i in 77% yield, mp 112–113 °C (EtOH). 1H NMR (CDCl3) δ 1.67–1.87 (m, 8H), 2.43–2.63 (m, 8H), 3.63 (s, 4H), 3.97 (s, 2H), 6.81 (s, 2H), 7.42 (d, J=8.6 Hz, 2H), 7.75 (d, J=8.7 Hz, 2H). Anal. (C23H30N3O3SCl) C, H, N, Cl.
N-[3,5-bis(1-pyrrolidylmethyl)-4-hydroxybenzyl]-4-acetamidobenzenesulfamide (6m)
N-(4-hydroxybenzyl)-4-acetamidobenzenesulfamide (13) was obtained in 81% yield, mp 185–186 °C (EtOH). 1H NMR (CDCl3) δ 2.12 (s, 3H), 3.97 (s, 2H), 6.72 (d, J=8.7 Hz, 2H), 7.08 (d, J=8.7 Hz, 2H), 7.78 (m, 4H). Anal. (C15H16N2O4S) C, H, N.
Mannich reaction of 13 gave product 6m in 93% yield, mp 105–107 °C (EtOH-H2O). 1H NMR (CDCl3) δ 1.68–1.88 (m, 8H), 2.16 (s, 3H), 2.48–2.68 (m, 8H), 3.65 (s, 4H), 3.94 (s, 2H), 6.83 (s, 2H), 7.60 (d, J=8.8 Hz, 2H), 7.69 (d, J=8.8 Hz, 2H), 8.37 (s, 1H). Anal. (C25H34N4O4S·H2O) C, H, N.
N-[3,5-bis(1-pyrrolidylmethyl)-4-hydroxyphenyl]-N-methyl-4-methyl-benzenesulfamide (6c)
A mixture of 4-methylaminophenol sulfate (0.34 g, 2.0 mmol), triethylamine (0.56 mL, 4.0 mmol) and 4-toluenesulfonyl chloride (0.37 g, 2.0 mmol) in ethanol (5 mL) was stirred at room temperature overnight. The solvent was removed under reduced pressure, and the residue was treated with ether. The ether layer was successively washed with 2 N hydrochloric acid, a saturated solution of NaHCO3 and water. The organic layer was dried over anhydrous Na2SO4. After removal of the solvent, the residue was recrystallized from EtOH to give 0.43 g (77%) of N-(4-hydroxyphenyl)-N-methyl-4-methylbenzenesulfamide (14), mp 136–137 °C. 1H NMR (CDCl3) δ 2.35 (s, 3H), 3.04 (s, 3H), 6.64 (d, J=8.4 Hz, 2H), 6.82 (d, J=8.4 Hz, 2H), 7.14 (d, J=8.7 Hz, 2H), 7.36 (d, J=8.7 Hz, 2H). Anal. (C14H15NO3S) C, H, N.
A mixture of 14 (0.33 g, 1.2 mmol), pyrrolidine (0.33 mL, 4.0 mmol) and a 36%–38% solution of formaldehyde (0.33 mL, 4.4 mmol) in EtOH (3 mL) was refluxed for 4 h. The solvent was removed on a rotary evaporator, and the crude product was purified by column chromatography (silica gel, AcOEt:MeOH:25% NH4OH=9:1:0.05) to give an oil that crystallized upon standing. Recrystallization from EtOH-H2O gave 0.41 g (78%) of 6c, mp 78–79 °C. 1H NMR (CDCl3) δ 1.68–1.88 (m, 8H), 2.38 (s, 3H), 2.43–2.63 (m, 8H), 3.10 (s, 3H), 3.66 (s, 4H), 6.74 (s, 2H), 7.17 (d, J=8.8 Hz, 2H), 7.40 (d, J=8.8 Hz, 2H). Anal. (C24H33N3O3S) C, H, N.
N-[3,5-bis(1-pyrrolidylmethyl)-4-hydroxyphenyl]-N-methyl-4-methoxy-benzenesulfamide (6g)
In the same manner as for the preparation of 14 and 6c, N-(4-hydroxyphenyl)-N-methyl-4-methoxybenzenesulfamide (15) was obtained in 95% yield, mp 89–90 °C (EtOH). 1H NMR (CDCl3) δ 3.04 (s, 3H), 3.80 (s, 3H), 6.65 (d, J=8.4 Hz, 2H), 6.72–6.89 (m, 4H), 7.41 (d, J=8.7 Hz, 2H). Anal. (C14H15NO4S) C, H, N.
Mannich reaction of 15 gave the product 6g in 78% yield, mp 125–127 °C (EtOH-H2O). 1H NMR (CDCl3) δ 1.69–1.89 (m, 8H), 2.46–2.66 (m, 8H), 3.10 (s, 3H), 3.68 (s, 4H), 3.83 (s, 3H), 6.77 (s, 2H), 6.86 (d, J=8.7 Hz, 2H), 7.46 (d, J=8.7 Hz, 2H). Anal. (C24H33N3O4S) C, H, N.
N-[3,5-bis(1-pyrrolidylmethyl)-4-hydroxyphenyl]-N-methyl-4-chloro-benzenesulfamide (6j)
In the same manner as for the preparation of 14 and 6c, N-(4-hydroxyphenyl)-N-methyl-4-chlorobenzenesulfamide (16) was obtained in 80% yield, mp 162–163 °C (Et2O). 1H NMR (CDCl3) δ 3.16 (s, 3H), 6.73 (d, J=8.6 Hz, 2H), 6.89 (d, J=8.6 Hz, 2H), 7.55 (m, 4H). Anal. (C13H12NO3SCl) C, H, N, Cl.
Mannich reaction of 16 gave the product 6j in 78% yield, mp 87–88 °C (EtOH-H2O). 1H NMR (CDCl3) δ 1.69–1.89 (m, 8H), 2.42–2.62 (m, 8H), 3.12 (s, 3H), 3.65 (s, 4H), 6.73 (s, 2H), 7.37 (d, J=8.7 Hz, 2H), 7.46 (d, J=8.7 Hz, 2H). Anal. (C23H30N3O3SCl) C, H, N, Cl.
N-[3,5-bis(1-pyrrolidylmethyl)-4-hydroxyphenyl]-N-methyl-4-acetamido-benzenesulfamide (6n)
In the same manner as for the preparation of 14 and 6c, N-(4-hydroxyphenyl)-N-methyl-4-acetamidobenzenesulfamide (17) was obtained in 77% yield, mp 202–203 °C (EtOH). 1H NMR (CDCl3) δ 2.07 (s, 3H), 3.03 (s, 3H), 6.64 (d, J=8.4 Hz, 2H), 6.82 (d, J=8.4 Hz, 2H), 7.36 (d, J=8.8 Hz, 2H), 7.70 (d, J=8.8 Hz, 2H). Anal. (C15H16N2O4S) C, H, N.
Mannich reaction of 17 gave the product 6n in 81% yield, mp 96–97 °C (EtOH-H2O). 1H NMR (CDCl3) δ 1.67–1.87 (m, 8H), 2.17 (s, 3H), 2.44-2.64 (m, 8H), 3.09 (s, 3H), 3.64 (s, 4H), 6.73 (s, 2H), 7.43 (d, J=9.0 Hz, 2H), 7.57 (d, J=9.0 Hz, 2H), 8.20 (brs, 1H). Anal. (C25H34N4O4S ·0.5H2O) C, H, N.
N-[3,5-bis(1-pyrrolidylmethyl)-4-hydroxyphenyl]-N-ethyl-4-methyl-benzenesulfamide (6d)
Using the same procedure as for the preparation of 6a, compound 6d was prepared from 4-ethylamino-2,6-bis(1-pyrrolidyl methyl)phenol and 4-toluenesulfonyl chloride in 48% yield, mp 119–120 °C (AcOEt). 1H NMR (CDCl3) δ 1.21 (t, J=7.0 Hz, 3H), 1.60–2.12 (m, 8H), 2.51 (s, 3H), 2.68–3.20 (m, 10H), 3.85 (s, 4H), 6.92 (s, 2H), 7.40 (d, J=8.7 Hz, 2H), 7.77 (d, J=8.7 Hz, 2H). Anal. (C25H35N3O3S) C, H, N.
N-[3,5-bis(1-piperidylmethyl)-4-hydroxyphenyl]-1-naphthalenesulfamide (6p)
A mixture of N-(4-hydroxyphenyl)-1-naphthalenesulfonamide (2.0 g, 6.7 mmol), 37% aqueous formaldehyde (4.5 g, 56 mmol) and piperidine (5.6 g, 66 mmol) in ethanol (100 mL) was heated to reflux for 12 h. The ethanol was removed by evaporation under vacuum, and chloroform was added to the residue. The organic layer was washed with water and dried over anhydrous Na2SO4. The chloroform was then removed in vacuo, and the residue was triturated in water to give a solid, which was recrystallized from ethanol to give the title product 6p (1.4 g, 42%), mp 197–198 °C. 1H NMR (CDCl3) δ 1.35–1.50 (m, 12H), 2.10–2.21 (m, 8H), 3.28 (s, 4H), 6.45 (s, 2H), 7.24–8.04 (m, 6H), 8.56 (m, 1H). Anal. (C28H35N3O3S) C, H, N.
N-[3,5-bis(1-pyrroliylmethyl)-4-hydroxyphenyl]-1-naphthalenesulfamide (6o)
Compound 6o was prepared by the same procedure as that used for 6p in 34% yield, mp 181–182 °C (EtOH). 1H NMR (CDCl3) δ 1.52–1.80 (m, 8H), 2.10–2.20 (m, 8H), 3.42 (s, 4H), 6.45 (s, 2H), 7.25–8.00 (m, 6H), 8.55 (m, 1H). Anal. (C26H31N3O3S) C, H, N.
Author contribution
Dong-lu BAI, Wei-zhou CHEN, and Yi-ping WANG designed and supervised the research project; Yun-xin BO, Zhong-liang HU, and Ai-li KANG performed the chemical synthesis; Yue-li DONG, Wei-kang SUN, and Yi-ping WANG performed the animal assays; Wei WANG and Yi-ping WANG performed the assays in vitro; Dong-lu BAI and Yi-ping WANG analyzed the data and wrote the manuscript.
References
Echt DS, Liebson PR, Mitchell LB, Peters RW, Obias-Manno D, Barker AH, et al. Mortality and morbidity in patients receiving encainide, flecainide, or placebo. The Cardiac Arrhythmia Suppression Trial. N Engl J Med 1991; 324: 781–8.
Nattel S, Carlsson L . Innovative approaches to anti-arrhythmic drug therapy. Nat Rev Drug Discov 2006; 5: 1034–49.
Walker MJA, So PPS . Anti-arrhythmics, In: Comprehensive Medicinal Chemistry II, Vol. 6 (Williams, M Ed). Amsterdam: Elsevier; 2007. p 729–62.
Li LQ, Qu ZX, Wang ZM, Zeng YL, Ding GS, Hu GJ, et al. Studies on a new antiarrhythmic drug changrolin — 4-(3′, 5′- bis(N-pyrrolidinylmethyl)-4′-hydroyanilino)quinazoline. Sci Sin 1979; 22: 1220–8.
Changrolin Coordination Research Group. Clinical observation on changrolin, an antiarrhythmic drug. Nat Med J Chin 1978; 58: 84–6.
Sun CJ, Zhang XY, Yang XZ, Wang PP, Shen J, Shu Y, et al. Studies on drugs for coronary diseases II. Synthesis of compounds related to changrolin as potential antiarrhythmic drugs. Yao Xue Xue Bao 1981; 16: 564–70.
Kang AL, Sun CJ . Studies on drugs for coronary diseases III. Synthesis of some Mannich lases of substituted aminophenols. Yao Xue Xue Bao 1986; 21: 892–8.
Chen WZ, Dong YL, Ding GS . Cardiovascular effect of a new antiarrhythmic drug — changrolin. Yao Xue Xue Bao 1979; 14: 710–4.
Stout DM, Matier WL, Barcelon-Yang C, Reynolds RD, Brown BS . Synthesis and antiarrhythmic and parasympatholytic properties of substituted phenols. 1. Heteroarylamine derivatives. J Med Chem 1983; 26: 808–13.
Stout DM, Matier WL, Barcelon-Yang C, Reynolds RD, Brown BS . Synthesis and antiarrhythmic and parasympatholytic properties of substituted phenols. 2. Amides. J Med Chem 1984; 27: 1347–50.
Stout DM, Matier WL, Barcelon-Yang C, Reynolds RD, Brown BS . Synthesis and antiarrhythmic and parasympatholytic properties of substituted phenols. 3. Modification of the linkage region. J Med Chem 1985; 28: 295–8.
Stout DM, Black LA, Barcelon-Yang C, Matier WL, Brown BS, Quon CY, et al. Ester derivatives of 2,6-bis(1-pyrrolidinylmethyl)-4-benzamidophenol as short-acting antiarrhythmic agents 1. J Med Chem 1989; 32: 1910–3.
Chorvat RJ, Black LA, Ranade VV, Barcelon-Yang C, Stout DM, Brown BS, et al. Mono- and bis(aminomethyl)phenylacetic acid esters as short-acting antiarrhythmic agents 2. J Med Chem 1993; 36: 2494–8.
Bo YX . The preparation of changrolin analogs [dissertation]. Shanghai Institute of Materia Medica, Chinese Academy of Sciences; 1991.
Chen WZ, Dong YL, Zhang YF, Ding GS . Antiarrhythmic effects of guan-fu base A. Zhongguo Yao Li Xue Bao 1983; 4: 247–50.
Dong YL, Chen WZ . Effects of guan-fu base A on experimental cardiac arrhythmia and myocardial contractility. Yao Xue Xue Bao 1995; 30: 577–82.
Kuang Y, Liu TP . Rate-dependent depression of maximal rate of depolarization in guinea pig papillary muscle action potentials by changrolin. Zhongguo Yao Li Xue Bao 1990; 11: 225–9.
Farmer BB, Mancina M, Williams ES, Watanabe AM . Isolation of calcium tolerant myocytes from adult rat heart: review of the literature and description of a method. Life Sci 1983; 33: 1–18.
Wang W, Hu GY, Wang YP . Selective modulation of L-type calcium current by magnesium lithospermate B in guinea-pig ventricular myocytes. Life Sci 2006; 78: 2989–97.
Wang W . Electrophysiological characterization of new cardiovascularly active compounds and cholinesterase inhibitors in guinea-pig heart [dissertation]. Shanghai Institute of Materia Medica, Chinese Academy of Sciences; 2005.
Acknowledgements
This work was supported by a grant from the Shanghai Committee of Science and Technology, China (No 975419000-4), and the Ministry of Science and Technology of China (Nos 2002AA2Z3123, 2005AA2Z3D60, and 2008ZX09101-004).
This paper is dedicated to the 80th anniversary of the founding of Shanghai Institute of Materia Medica, Chinese Academy of Sciences.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Supplementary information (IR spectra and elemental analysis data for all new compounds) are available at website of Acta Pharmacologica Sinica on NPG.
Supplementary information
Appendix
Data of the IR spectra and elemental analysis of all new compounds. (DOC 120 kb)
Rights and permissions
About this article
Cite this article
Bai, Dl., Chen, Wz., Bo, Yx. et al. Discovery of N-(3,5-bis(1-pyrrolidylmethyl)-4-hydroxybenzyl)-4-methoxybenzenesulfamide (sulcardine) as a novel anti-arrhythmic agent. Acta Pharmacol Sin 33, 1176–1186 (2012). https://doi.org/10.1038/aps.2012.119
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2012.119
Keywords
This article is cited by
-
RETRACTED ARTICLE: Characterization of Preclinical Pharmacokinetic Properties and Prediction of Human PK Using a Physiologically Based Pharmacokinetic Model for a Novel Anti-Arrhythmic Agent Sulcardine Sulfate
Pharmaceutical Research (2021)
-
Barbaloin inhibits ventricular arrhythmias in rabbits by modulating voltage-gated ion channels
Acta Pharmacologica Sinica (2018)
-
Multiple Dose Pharmacokinetics and Safety of Sulcardine Sulfate in Healthy Chinese Male Subjects: An Open-Label Phase I Clinical Study
European Journal of Drug Metabolism and Pharmacokinetics (2017)
-
Oral Bioavailability and Mass Balance Studies of a Novel Anti-arrhythmic Agent Sulcardine Sulfate in Sprague–Dawley Rats and Beagle Dogs
European Journal of Drug Metabolism and Pharmacokinetics (2017)
-
Celebrating the 80th anniversary of the Shanghai Institute of Materia Medica, Chinese Academy of Sciences (SIMM)
Acta Pharmacologica Sinica (2012)
