
Abstract
Aim:
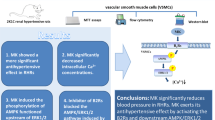
To identify a key protein that binds monomeric G protein RhoA and activates the RhoA/Rho kinase/MYPT1 axis in vascular smooth muscle cells (VSMCs) upon angiotensin II (Ang II) stimulation.
Methods:
Primary cultured VSMCs from Sprague-Dawley rats were transfected with siRNAs against leukemia-associated RhoGEF (LARG), and then treated with Ang II, losartan, PD123319, or Val5-Ang II. The target mRNA and protein levels were determined using qPCR and Western blot analysis, respectively. Rat aortic rings were isolated, and the isometric contraction was measured with a force transducer and recorder.
Results:
Stimulation with Ang II (0.1 μmol/L) for 0.5 h significantly increased the level of LARG mRNA in VSMCs. At 3, 6, and 9 h after the treatment with Ang II (0.1 μmol/L) plus AT2 antagonist PD123319 (1 μmol/L) or with AT1 agonist Val5-Ang II (1 μmol/L), the LARG protein, RhoA activity, and phosphorylation level of myosin phosphatase target subunit 1 (MYPT1) in VSMCs were significantly increased. Knockdown of LARG with siRNA reduced these effects caused by AT1 receptor activation. In rat aortic rings pretreated with LARG siRNA, Ang II-induced contraction was diminished.
Conclusion:
Ang II upregulates LARG gene expression and activates the LARG/RhoA/MYPT1 axis via AT1, thereby maintaining vascular tone.
Similar content being viewed by others


Introduction
Angiotensin II (Ang II) plays a pivotal role in blood pressure regulation. Activation of Ang II receptors, which increases free intracellular Ca2+ concentrations and increases myosin light chain (MLC) kinase activity, can cause vessel constriction. These events result in MLC phosphorylation and subsequent smooth muscle contraction1,2,3,4. However, a portion of the agonist-induced smooth muscle contraction, termed Ca2+ sensitization, is mediated by the Ca2+-independent activation of monomeric G protein RhoA and the downstream target Rho-kinase4,5.
RhoA activity is regulated by Rho guanine nucleotide exchange factors (RhoGEFs) and Rho GDP dissociation inhibitors6,7,8,9. Despite RhoA's requirement of basal activity for homeostatic functions under physiological conditions, sustained- and over-activation of RhoA may result in pathological effects, including long-lasting contractions in vascular smooth muscle cells (VSMCs)10.
A link between Ang II type 1 receptors (AT1) and the RhoA/Rho kinase pathway has been reported4,11. Three RhoGEFs may link AT1 to RhoA activation: PSD-95/Disc-large/ZO-1 homology (PDZ)-RhoGEF, leukemia-associated RhoGEF (LARG), and p115-RhoGEF4,12,13. It has been reported that Ang II transiently upregulates the expression of LARG mRNA and protein, but not PDZ-RhoGEF or p115-RhoGEF in a rat VSMC model14. A later study also found that the overexpression of PDZ-RhoGEF, but not LARG or p115-RhoGEF, enhances Ang II-stimulated RhoA activity, and the knockdown of PDZ-RhoGEF decreases Ang II-stimulated RhoA activity in VSMCs15. Dubash et al showed that the knockdown of both p115-RhoGEF and LARG significantly decreased both RhoA activation and the formation of stress fibers and also decreased focal adhesions downstream of fibronectin adhesions16. Wirth et al established the role of LARG in physiological responses to Ang II stimulation by showing that arteries in LARG knockout mice produce a reduced maximum degree of contraction in response to Ang II and fail to develop deoxycorticosterone acetate plus NaCl (DOCA/salt)-induced hypertension17. Another study reported that p115-RhoGEF is responsible for AT1-mediated RhoA activation and that ablation of the p115-RhoGEF gene in VSMCs prevents Ang II-induced hypertension8. All of these previous results are inconsistent with RhoGEF playing a primary role in the regulation of RhoA/Rho-kinase.
Rho-kinase is a divergent point for multiple signals in the RhoA/Rho-kinase pathway for the regulation of smooth muscle cell function. Rho-kinase increases MLC phosphorylation via the phosphorylation of myosin phosphatase target subunit 1 (MYPT1) on MLCP. Therefore, MYPT1 may play a critical role in the Ca2+ sensitization of myofilaments and in maintaining vascular smooth muscle tension under constant Ca2+ concentration. Nevertheless, the role of LARG gene expression in MYPT1 phosphorylation and Ca2+ sensitization of VSMCs is not yet understood.
The objective of this study was to identify a specific molecule(s) that activates RhoA in response to stimulation by an AT1 agonist in primary cultured VSMCs. Additionally, we investigated the association between LARG gene expression and the extent of MYPT1 phosphorylation in VSMCs after AT1 receptor activation. Finally, we used LARG siRNA knockdown to study aortic ring contractile forces ex vivo.
Materials and methods
Cell culture
Sprague-Dawley (SD) rat aorta smooth muscle cells (RAOSMC)6 were obtained from the tunica intima and the inner layer of tunica media of healthy and fibrous plaque-free rat aorta. The cells were cultured in M199 culture medium, supplemented with 20% fetal bovine serum10, penicillin (100 U/mL), and streptomycin (100 μg/mL) at 37 °C in a humidified atmosphere of 95% air and 5% CO2. The cells were passaged twice a week by harvesting with trypsin and seeding at a 1:4 ratio into 100-mm dishes. The cells from passage two through passage five were used in all of the experiments. The cells were harvested at the indicated times and used for qPCR or Western blot analysis.
Isolation of total RNA and reverse transcription
RNA from the cultured cells was isolated using the TRIzol reagent10. All reverse-transcriptase reactions were performed using a High-Capacity cDNA Reverse Transcription kit according to the manufacturer's instructions2. The cDNA was used immediately for qPCR or stored at −80 °C.
qPCR assay
We performed the qPCR assays using a 7000 Real-time PCR Sequence Detection System2. The cDNA generated from total RNA (30 ng) was used for each reaction. The amount of each target mRNA relative to the internal normalization control, GAPDH, was calculated using the Comparative Ct method as follows:

Where,

ΔCt, exp (the change in cycle threshold) is the target mRNA's Ct value in the experimental group (target gene; treated duration of 0.5, 3, or 6 h), and ΔCt, con is the Ct value of the target mRNA in the control group (target gene; 0 h; non-stimulated conditions).
Preparation of cell extracts and Western blot analysis
To prepare whole-cell lysates and to measure membrane translocation of RhoA (activation of RhoA), culture dishes were cooled on ice and washed twice with ice-cold phosphate-buffered saline (PBS). All procedures were performed on ice. Lysis buffer for whole-cell lysates [cold PBS solution (pH 7.4) containing 1 mmol/L ethylenediamine tetraacetic acid (EDTA), 25 mmol/L HEPES, 150 mmol/L NaCl, 10 mmol/L MgCl2, 10% glycerol, and 1% Igepal CA-630] and for RhoA translocation detection [cold PBS solution (pH 7.4) containing 2 mmol/L EDTA, 2 mmol/L phenylmethylsulfonyl fluoride (PMSF), and 0.8 μg/mL leupeptin] was added, and the plates were placed on ice for 2 min. The cells were scraped off and transferred to Eppendorf tubes.
For whole-cell lysate preparation, the samples were sonicated for 5 s and then centrifuged at 12 000×g for 15 min at 4 °C. The supernatant was transferred to a new tube and kept on ice. For RhoA translocation detection, the samples were centrifuged at 500×g for 10 min at 4 °C to remove the nuclei. The membrane and cytosolic fractions were further separated by centrifugation at 100 000×g for 1 h at 4 °C4. The supernatant (cytoplasmic fraction) was kept on ice, and the pellet (membrane fraction) was resuspended in PBS buffer containing Tris-HCl, 100 mmol/L NaCl, 1% Triton X-100, 0.1% sodium dodecyl sulfate (SDS), 2 mmol/L EDTA, 2 mmol/L PMSF, and 0.8 μg/mL leupeptin. The protein concentration was determined using a BSA Protein Assay kit13.
Protein (40 μg) was separated by electrophoresis on a 12% or 6% polyacrylamide gel and transferred to a polyvinylidene difluoride membrane. Nonspecific binding sites were blocked using Protein-Free Blocking Buffers13 for 1 h at 24 °C. The membranes were then incubated overnight at 4°C with the following antibodies: anti-RhoA (1:1000)14, anti-flotillin-2 (1: 1000)14, anti-LARG (1:1000)14, anti-phospho-MYPT1 (Thr696) (1:2000)11, anti-total-MYPT1 (1:2000)5, anti-tubulin (1:2000)14, or anti-GAPDH (1:1000)7. Following exposure to a horseradish peroxidase-conjugated secondary anti-mouse, anti-rabbit, or anti-goat antibody, the membrane was subjected to the ECL reagent11 and exposed to film18. The bands were quantified using AlphaImage software3.
Rho GTPase activity assay
RhoA activity was determined using an absorbance based G-LISA™ RhoA activation assay kit (Cat#BK124)8, according to the manufacturer's instructions15. This assay employs a Rho-GTP binding protein, which coats the wells of a 96-well plate. Active, GTP-bound Rho in the cell lysates will bind to the wells, while inactive GDP-bound Rho is removed in the washing steps. The bound active RhoA is detected by incubation with a specific RhoA antibody followed by an HRP-conjugated secondary antibody and a detection reagent, after which the absorbance is read on a SpectraMax M5 Microplate Reader12. Primary VSMCs were cultured in 100-mm dishes and treated as indicated above. Proteins were harvested by incubating the cell with the provided cell lysis buffer with protease inhibitors and centrifugating at 12 000×g at 4 °C for 2 min to remove cell debris. The protein concentration was determined according to the manufacturer's protocol, and the cell extracts were equalized to a protein concentration of 1.5 mg/mL for assay. Absorbance was detected according to the manufacturer's recommendation after a 45-min incubation at room temperature with the primary anti-RhoA antibody19.
siRNA experiments
Scrambled siRNA (5 nmol/L) or siRNA against LARG (5 nmol/L) was introduced into the VSMCs using the siPORTTM NeoFXTM Transfection Agent (Cat#AM4511)1. LARG siRNA1 (Cat#4390771) sequences were as follows: sense, 5′-GCAUCGAUCUGUAUACACUtt-3′, and antisense, 5′-AGUGUAUACAGAUCGAUGCtt-3′ the scrambled siRNA (Cat#4390843) was an siRNA sequence not homologous to any known gene1. After transfection for 36 h, the cells were treated with Ang II, losartan, PD123319, or Val5-Ang II, which were dissolved in distilled water. Cells were harvested at the indicated times and used for qPCR or Western blot analysis.
Tissue preparation and isometric force recording
All animal procedures were performed in accordance with the Institutional Animal Care and Use Committee of National Taiwan University College of Medicine (Approval No 20090097). Male SD rats (275–300 g) were purchased from the National Laboratory Animal Centre of Taiwan, China. The rats were anesthetized with sodium pentobarbital (50 mg/kg, ip), and the thoracic aorta was excised and cleaned of adventitial tissue using a physiological saline solution (Krebs-Henseleit solution, KHS, pH 7.4). The aortas were cut into 3 mm rings, and the endothelia were removed.
The rings were incubated in culture medium containing either an siRNA against LARG (10 nmol/L) or scrambled siRNA (10 nmol/L) for 48 h at 37 °C20,21. The siRNA was introduced using the siPORTTM NeoFXTM Transfection Agent. After incubation, the aortic rings were mounted in myograph organ baths containing KHS at 37 °C and gassed with 95% O2 and 5% CO2. The isometric force generation was recorded with a force transducer (TypeBG25) and recorder (RS 3400)9. The rings were placed under 15 mN tension and equilibrated for 120 min. During this period, KHS was refreshed every 30 min, and the rings were re-stretched as necessary to maintain 15 mN tension. After equilibration, the rings were contracted using phenylephrine (PE, 0.1 μmol/L). Acetylcholine (ACh, 1 μmol/L) was added during the contraction plateau phase to verify the efficient disruption of the endothelium. The drugs and reagents were washed out until the contractile response returned to baseline levels. We then recorded the contractile response curves for Ang II, losartan, and PD123319, which were dissolved in distilled water22,23.
Statistical analysis
The data are expressed as the mean±SD, and n represents the number of independent experiments. A t-test, ANOVA and the post-hoc test (Tukey) were employed to analyze the results. A P-value of less than 0.05 was considered significant. All statistical analyses were performed using SPSS19.015.
Manufacturer reference guide
-
1)
Ambion, Austin, TX, USA
-
2)
Applied Biosystems, Foster City, CA, USA
-
3)
Alpha Innotech Corporation, San Leandro, CA, USA
-
4)
Beckman Instruments, Fullerton, CA, USA
-
5)
BD Biosciences Pharmingen, San Diego, CA, USA
-
6)
Cell Applications, San Diego, CA, USA
-
7)
Chemicon, Temecula, CA, USA
-
8)
Cytoskeleton, Denver, CO, USA
-
9)
Gould Inc Cleveland, OH, USA
-
10)
Invitrogen, Carlsbad, CA, USA
-
11)
Millipore Corporation, Billerica, MA, USA
-
12)
Molecular Devices, Sunnyvale, CA, USA
-
13)
Pierce Chemical, Rockford, IL, USA
-
14)
Santa Cruz Biotechnology, Santa Cruz, CA, USA
-
15)
SPSS, Inc, Chicago, IL, USA
Results
RhoGEFs gene expression after Ang II stimulation of rat aortic VSMCs
We examined mRNA transcription by qPCR following stimulation with Ang II (0.1 μmol/L) for 0.5, 3, or 6 h. The results demonstrated that LARG mRNA significantly increased at 0.5 h post Ang II stimulation (n=6) (Figure 1A), whereas the mRNA levels of PDZ-RhoGEF and p115-RhoGEF did not show any significant change after stimulation (n=6) (Figure 1B, 1C).

Expression of RhoGEFs mRNA in rat VSMCs after Ang II stimulation. VSMCs were treated with or without 0.1 μmol/L Ang II for 0.5, 3, or 6 h. We measured mRNA for (A) LARG, (B) PDZ-RhoGEF, and (C) p115-RhoGEF by qPCR and normalized to GAPDH. Data are presented as the mean±SD (n=6). The white bars indicate the unstimulated control. cP<0.01 vs control group.
LARG is induced by Ang II through AT1
The activation of AT1, by the agonist Val5-Ang II (Val5) (1 μmol/L), significantly induced LARG protein expression (n=6) after a 3 h drug treatment (Figure 2A). Ang II (0.1 μmol/L) plus PD123319 (1 μmol/L), an AT2 antagonist, also increased LARG protein levels (n=6) after the 3 h treatment (Figure 2B). The elevation in LARG protein levels after AT1 activation was maintained for 9 h (Figure 2A, 2B). Losartan (1 μmol/L), an AT1 antagonist, prevented Ang II stimulated LARG expression (n=6) (Figure 2C).

Expression of LARG protein in rat VSMCs following various treatments. LARG protein expression was observed at the indicated times after the addition of (A) AT1 agonist Val5-Ang II (Val5) (1 μmol/L), (B) Ang II (0.1 μmol/L) together with the AT2 antagonist, PD123319 (1 μmol/L), or (C) Ang II (0.1 μmol/L) plus the AT1 antagonist losartan (1 μmol/L). Western blot analysis was used to determine protein levels, and GAPDH was used as the internal control. Results are presented as the mean±SD (n=6). The white bars indicate the unstimulated control. Western blot images are from representative experiments. cP<0.01 vs control group.
AT1 stimulation increased RhoA activity and translocation to the cell membrane
Once activated, RhoA partially translocates from the soluble (cytosolic) fraction to the particulate (membrane) fraction24,25. RhoA activation is described by the ratio of RhoA in the particulate versus that in the soluble fraction. VSMCs were stimulated with Val5 (1 μmol/L) for 3, 6, or 9 h. Figure 3A shows that RhoA activity continuously increased over 9 h (n=6), while Figure 3B shows that RhoA activity was also enhanced following treatment with Ang II (0.1 μmol/L) plus PD123319 (1 μmol/L) (n=6). Treatment with losartan (1 μmol/L) blocked the anticipated enhancement of RhoA translocation in response to stimulation with Ang II (n=6) (Figure 3C).

(A–C) Translocation of RhoA (active RhoA) after AT1 activation in rat VSMCs. VSMCs were stimulated with or without (A) Val5 (1 μmol/L), (B) Ang II (0.1 μmol/L) together with PD123319 (1 μmol/L), or (C) Ang II (0.1 μmol/L) plus losartan (1 μmol/L) for the indicated times. RhoA translocation (RhoA activity) was determined by Western blot analysis and presented as the ratio of protein in the particulate (membrane) fraction vs the soluble (cytosolic) fraction. The white bars indicate the unstimulated control. (D–F) Expression of LARG mRNA and protein and RhoA activity after LARG knockdown. VSMCs were treated with scrambled siRNA or LARG siRNA for 48 h. Subsequently, the cells were stimulated with (D) Ang II (0.1 μmol/L) for 0.5 h, and the analysis of LARG mRNA was performed by qPCR. The cells were stimulated with 1 μmol/L Val5 for 3 h and (E) LARG protein was measured by Western blot analysis, or (F) RhoA activity was measured by the G-LISA kit. LARG mRNA and protein expression were normalized to GAPDH and reported relative to a negative control (scrambled siRNA and without AT1 agonists stimulation). Results are presented as the mean±SD (n=6). Western blot images are from a representative experiment. Flotillin-2, a positive control of the plasma membrane fraction, was used to confirm the non-contaminated fraction. bP<0.05, cP<0.01 vs the control group. fP<0.01 vs the scrambled siRNA stimulated by the AT1 agonists group.
Knockdown of LARG decreased RhoA activity
To determine whether LARG gene expression was associated with RhoA activity, we knocked down LARG using siRNA. As expected, LARG mRNA was significantly reduced in the LARG siRNA pretreatment group and in the LARG siRNA pretreatment plus Ang II stimulation (0.1 μmol/L) group (n=6) (Figure 3D). LARG proteins were also significantly decreased in the LARG siRNA pretreatment group and in the LARG siRNA plus Val5 (1 μmol/L) group (n=6) (Figure 3E). The effect of LARG knockdown on RhoA activity was assessed by an absorbance-based G-LISA™ assay kit. RhoA activity was significantly suppressed in the LARG siRNA pretreatment group and in the LARG siRNA plus Val5 group (n=6) (Figure 3F). This finding is consistent with the hypothesis that LARG acts upstream of the RhoA signaling pathway.
AT1 activation enhanced MYPT1 phosphorylation
The phosphorylation of MYPT1 at Thr696 (phospho-MYPT1) in VSMCs was measured prior to stimulation (0 h group) and after stimulation with Val5 (1 μmol/L) or Ang II (0.1 μmol/L) plus PD123319 (1 μmol/L) at 3, 6, and 9 h (Figure 4A & 4B). The results showed that MYPT1 phosphorylation was enhanced after AT1 activation, with the phosphorylation of MYPT1 consistently increasing over 9 h (n=6).

(A, B) Levels of MYPT1 phosphorylation after AT1 activation in rat VSMCs. VSMCs were stimulated with or without (A) Val5 (1 μmol/L) or (B) Ang II (0.1 μmol/L) together with PD123319 (1 μmol/L) for 3, 6, or 9 h. Protein levels of phosphorylated-MYPT1 were determined by Western blot analysis. The white bars indicate the unstimulated control. (C) The phosphorylation levels of MYPT1 in rat VSMCs, after knockdown of LARG, were determined by Western blot analysis. GAPDH was used as the internal control. Data are reported relative to a negative control (scrambled siRNA and without Val5 stimulation). The Western blot images are from a representative experiment. Results are presented as the mean±SD (n=6). bP<0.05, cP<0.01 vs control group. fP<0.01 vs the scramble siRNA stimulated by the Val5 group.
Knockdown of LARG decreases MYPT1 phosphorylation
LARG knockdown was utilized to determine whether LARG gene expression is associated with MYPT1 phosphorylation. Rat VSMCs were transfected with LARG siRNA or scrambled siRNA. After 36 h, the cells were treated with or without Val5 (1 μmol/L) for three or nine hours and harvested. LARG silenced cells showed a markedly decreased MYPT1 phosphorylation compared to the scrambled siRNA-treated control. Treatment with Val5 (1 μmol/L) did not alter the MYPT1 phosphorylation levels (Figure 4C). These results suggest that LARG gene expression regulates MYPT1 phosphorylation and Ca2+ sensitization of myofilaments in VSMCs in response to AT1 activation (n=6).
Pretreatment with LARG siRNA diminishes Ang II-induced vasoconstriction
Aortic rings were transfected with scrambled siRNA or LARG siRNA for 48 h. Protein was extracted from each rat aortic ring and subjected to Western blot analysis to verify the efficiency of LARG knockdown (Figure 5A). The remaining rings were used to record contractile tension forces. Figure 5B1 shows a typical curve for Ang II-induced contraction of rat aortic rings. The antagonists evaluated included the AT2 antagonist PD123319 (1 μmol/L) and the AT1 antagonist losartan (1 μmol/L), as shown in Figure 5B2 and 5B3, respectively. We examined the effects of siRNA against LARG on the contraction of rat aortic rings in response to Ang II to clarify the role of LARG in Ang II/Ca2+-sensitization signaling of a large conduit artery (Figure 5B4). We found that the force of contraction was weakened in the LARG siRNA pretreated aortic rings in response to Ang II treatment compared to the contractile force of the scrambled-siRNA-treated controls [46.9%±0.8% vs 17.3%±2.1%, normalized to phenylephrine (PE)-induced maximal contraction, n=4, P<0.01; Figures 5B1, 5B4, and 5C]. The administration of losartan reduced the force associated with Ang II stimulation (46.9%±0.8% vs 16.1%±3.9%, normalized to PE-induced maximum contraction, n=4, P<0.01; Figure 5B1, 5B3, and 5C). After completion of the contractile tension recording experiments, the aortic rings were subjected to protein extraction and Western blot analysis to re-confirm the effect of LARG knockdown (Figure 5D).

Effects of PD123319 (1 μmol/L), losartan (1 μmol/L), or LARG siRNA on Ang II-induced contraction of rat aortic rings. (A) Before the contraction recordings were obtained, protein was extracted from the endothelium-removed aortic segments that were treated with scrambled siRNA or LARG siRNA for 48 h. LARG protein was measured by Western blot. GAPDH was used as an internal control. (B1–B4) Typical recordings of the denuded aortic rings following the treatments. (C) The magnitude of the contraction is expressed as a percentage of maximal phenylephrine (PE, 1 μmol/L)-induced contraction. (D) After recording, protein was extracted from the aortic rings, and Western blot analysis was performed. LARG protein expression was normalized to GAPDH. The aortic rings treated with scrambled siRNA and without antagonists were the control. Data are expressed as the mean±SD (n=4). cP<0.01 vs the control group.
Discussion
This study provides direct evidence that the activation of AT1 enhances LARG gene expression, which increases RhoA activity and MYPT1 phosphorylation, leading to inactivation of MLCP and the promotion of Ca2+ sensitization in VSMCs. These effects result in VSMCs contraction and vasoconstriction. Knockdown of LARG by siRNA decreased aortic ring contraction in response to Ang II stimulation. The results confirm that MLCP phosphorylation is directly regulated by LARG gene expression that occurs in response to AT1 activation.
We found that LARG acts upstream of the RhoA/Rho kinase pathway. By contrast, p115-RhoGEF and PDZ-RhoGEF, which exhibit trends similar to LARG following AT1 activation, did not reach statistical significance. Thus, we examined studies to determine the role of LARG in this pathway. The difference between our results and those observed by other authors may have been due to the different materials used for the experiments26. Aortae are composed of multiple tissue types; in addition to smooth muscle, they also contain connective tissue and fibroblasts. In this study, we used primary cultured VSMCs to eliminate the interference of signaling from different cell types. Ying et al showed that Ang II transiently upregulates LARG mRNA and increases Y27632-induced vasodilation in Ang II-incubated rat aortic rings14. Our findings support Ying's results.
VSMC contraction is dependent on the extent of MLC phosphorylation, which is regulated by the balance between Ca2+-dependent MLCK and Ca2+-independent MLCP activities7,27. Therefore, the regulation of MLCP activity by the phosphorylation of MYPT1 plays a critical role in Ang II-induced, Ca2+-independent, Ca2+ sensitization of VSMCs. Most studies investigating the calcium sensitization pathway in VSMCs either report RhoA activity or estimate vasorelaxation induced by Rho kinase inhibition14. However, Rho-kinase is a point of divergence for multiple signal pathways, and thus, Rho kinase mobility is not solely responsible for the Ca2+-sensitization of myofilaments. Because Rho-kinase induced phosphorylation of MLC occurs via phosphorylation of the MYPT1 on MLCP, a better method to identify the Ca2+ sensitivity of VSMCs might be to assess the phosphorylation of MYPT1.
Guilluy et al showed that MYPT1 phosphorylation levels are decreased in the aortic smooth muscle cells in p115-RhoGEF deficient mice treated with Ang II8. Another study showed that the Gα12Gα13/LARG pathway is crucial to saltinduced hypertension17. However, it has not been directly demonstrated that AT1 activated LARG gene expression regulates the phosphorylation of MYPT-1. Our results illustrate that the increased LARG protein expression that results from AT1 activation contributes to the enhancement of MYPT1 phosphorylation. Our ex vivo data also indicate that the specific knockdown of LARG in aortic rings results in a diminished contractile force in response to Ang II. Hypertension increases arterial tone in both small resistance arteries (myogenic tone) and large conduit arteries (spontaneous tone). It has also been established that RhoA signaling mediates the development of spontaneous tone in Ang II-induced hypertensive rats4. Our current findings may aid the elucidation of the mechanism involved in controlling spontaneous tone in the vascular wall of large conduit arteries.
The transient increase of LARG mRNA at 0.5 h in response to Ang II stimulation and its subsequent disappearance after 3 h is due to the short duration of Ang II stimulation and the decay of Ang II. In addition, LARG mRNA may be digested by RNase activity. LARG mRNA expression, in vivo, may be related to the continuous stimulation of intravascular Ang II, which is different from our in vitro observations. The physiological relevance of LARG expression is to maintain vascular tone by causing sustained contraction.
Our study has some limitations. Ang II-induced VSMC contraction is both calcium dependent and calcium independent (calcium sensitization). We use the same concentration of Ang II to stimulate VSMC in all of the independent experiments. Thus, the calcium-dependent pathway is purported to function the same when contributing to VSMC contraction. We only focused our studies on the calcium sensitization pathway, and we did not measure the intracellular calcium concentration. Protein kinase C (PKC) and its downstream target CPI-17, a 17 kDa PKC-potentiated inhibitor protein, is another potential mediator of Ca2+ sensitization. The activation of CPI-17, via the phosphorylation by PKC, inhibits the MLCP catalytic subunit PP1C27,28. We did not study the role of PKC or CPI-17 in LARG expression or the association of LARG protein with Ca2+ sensitization in VSMCs. These topics warrant further investigation.
In conclusion, the LARG/RhoA-Rho kinase/MYPT1 signaling pathway is identified as the predominant pathway involved in the Ca2+ sensitization of myofilaments. This signaling pathway responds to AT1-induced Ca2+ sensitivity in VSMCs. Stimulation of AT1 increases LARG protein expression, the extent of membrane localization of RhoA (active RhoA), and the phosphorylation of MYPT1.
Author contribution
Wei-chiao CHIU, Chia-ti TSAI, and Fu-tien CHIANG designed the research method; Wei-chiao CHIU, Chow-kai WU, and Shen-nan CHANG performed the research; Wei-chiao CHIU, Jyh-ming JUANG, Yung-zu TSENG, and Fu-tien CHIANG analyzed the data; and Wei-chiao CHIU, Yung-zu TSENG, and Fu-tien CHIANG wrote the paper.
References
Touyz RM, Schiffrin EL . Signal transduction mechanisms mediating the physiological and pathophysiological actions of angiotensin II in vascular smooth muscle cells. Pharmacol Rev 2000; 52: 639–72.
Touyz RM . The role of angiotensin II in regulating vascular structural and functional changes in hypertension. Curr Hypertens Rep 2003; 5: 155–64.
Kim S, Iwao H . Molecular and cellular mechanisms of angiotensin II-mediated cardiovascular and renal diseases. Pharmacol Rev 2000; 52: 11–34.
Jin L, Ying Z, Hilgers RH, Yin J, Zhao X, Imig JD, et al. Increased RhoA/Rho-kinase signaling mediates spontaneous tone in aorta from angiotensin II-induced hypertensive rats. J Pharmacol Exp Ther 2006; 318: 288–95.
Uehata M, Ishizaki T, Satoh H, Ono T, Kawahara T, Morishita T, et al. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature 1997; 389: 990–4.
Pagnin E, Semplicini A, Sartori M, Pessina AC, Calo LA . Reduced mRNA and protein content of rho guanine nucleotide exchange factor (RhoGEF) in Bartter's and Gitelman's syndromes: relevance for the pathophysiology of hypertension. Am J Hypertens 2005; 18: 1200–5.
Somlyo AP, Somlyo AV . Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol Rev 2003; 83: 1325–58.
Guilluy C, Bregeon J, Toumaniantz G, Rolli-Derkinderen M, Retailleau K, Loufrani L, et al. The Rho exchange factor Arhgef1 mediates the effects of angiotensin II on vascular tone and blood pressure. Nat Med 2010; 16: 183–90.
Shichrur K, Yalovsky S . Turning ON the switch — RhoGEFs in plants. Trends Plant Sci 2006; 11: 57–9.
Loirand G, Guilluy C, Pacaud P . Regulation of Rho proteins by phosphorylation in the cardiovascular system. Trends Cardiovasc Med 2006; 16: 199–204.
Maguire JJ, Davenport AP . Regulation of vascular reactivity by established and emerging GPCRs. Trends Pharmacol Sci 2005; 26: 448–54.
Banerjee J, Wedegaertner PB . Identification of a novel sequence in PDZ-RhoGEF that mediates interaction with the actin cytoskeleton. Mol Biol Cell 2004; 15: 1760–75.
Rossman KL, Der CJ, Sondek J . GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol 2005; 6: 167–80.
Ying Z, Jin L, Palmer T, Webb RC . Angiotensin II up-regulates the leukemia-associated Rho guanine nucleotide exchange factor (RhoGEF), a regulator of G protein signaling domain-containing RhoGEF, in vascular smooth muscle cells. Mol Pharmacol 2006; 69: 932–40.
Ying Z, Giachini FR, Tostes RC, Webb RC . PYK2/PDZ-RhoGEF links Ca2+ signaling to RhoA. Arterioscler Thromb Vasc Biol 2009; 29: 1657–63.
Dubash AD, Wennerberg K, Garcia-Mata R, Menold MM, Arthur WT, Burridge K . A novel role for Lsc/p115 RhoGEF and LARG in regulating RhoA activity downstream of adhesion to fibronectin. J Cell Sci 2007; 120: 3989–98.
Wirth A, Benyo Z, Lukasova M, Leutgeb B, Wettschureck N, Gorbey S, et al. G(12)-G(13)-LARG-mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat Med 2008; 14: 64–8.
Savoia C, Tabet F, Yao G, Schiffrin EL, Touyz RM . Negative regulation of RhoA/Rho kinase by angiotensin II type 2 receptor in vascular smooth muscle cells: role in angiotensin II-induced vasodilation in stroke-prone spontaneously hypertensive rats. J Hypertens 2005; 23: 1037–45.
Wang JP, Liu HX, Chen B, Li Q, Huang XL, Wang LQ, et al. RhoA/ROCK-dependent moesin phosphorylation regulates AGE-induced endothelial cellular response. Cardiovasc Diabetol 2012; 11: 7.
Sun D, Yan CD, Jacobson A, Jiang H, Carroll MA, Huang A . Contribution of epoxyeicosatrienoic acids to flow-induced dilation in arteries of male ER alpha knockout mice: role of aromatase. Am J Physiol Regul Integr Comp Physiol 2007; 293: R1239–46.
Guilluy C, Rolli-Derkinderen M, Loufrani L, Bourge A, Henrion D, Sabourin L, et al. Ste20-related kinase SLK phosphorylates Ser188 of RhoA to induce vasodilation in response to angiotensin II Type 2 receptor activation. Circ Res 2008; 102: 1265–74.
Ying Z, Giachini FR, Tostes RC, Webb RC . Salicylates dilate blood vessels through inhibiting PYK2-mediated RhoA/Rho-kinase activation. Cardiovasc Res 2009; 83: 155–62.
Lodi F, Cogolludo A, Duarte J, Moreno L, Coviello A, Peral De Bruno M, et al. Increased NADPH oxidase activity mediates spontaneous aortic tone in genetically hypertensive rats. Eur J Pharmacol 2006; 544: 97–103.
Bokoch GM, Bohl BP, Chuang TH . Guanine nucleotide exchange regulates membrane translocation of Rac/Rho GTP-binding proteins. J Biol Chem 1994; 269: 31674–9.
Yamakawa T, Tanaka S, Numaguchi K, Yamakawa Y, Motley ED, Ichihara S, et al. Involvement of Rho-kinase in angiotensin II-induced hypertrophy of rat vascular smooth muscle cells. Hypertension 2000; 35: 313–8.
Ying Z, Jin L, Dorrance AM, Webb RC . Increaseed expression of mRNA for regulator of G protein signaling domain-containing Rho guanine nucleotide exchange factors in aorta from stroke-prone spontaneously hypertensive rats. Am J Hypertens 2004; 17: 981–5.
Somlyo AP, Somlyo AV . Signal transduction by G-proteins, rho-kinase and protein phosphatase to smooth muscle and non-muscle myosin II. J Physiol 2000; 522: 177–85.
Somlyo AP, Somlyo AV . Signal transduction and regulation in smooth muscle. Nature 1994; 372: 231–6.
Acknowledgements
This study was supported by a grant (NSC98-2320-B002-035) from the National Science Council, Taiwan, China, and was supported in part by the Liver Disease Prevention & Treatment Research Foundation, Taiwan, China.
The authors acknowledge statistical assistance provided by the National Translational Medicine and Clinical Trial Resource Center (supported by the National Science Council of Taiwan, China; NSC101-2325-B-002-078) and the Department of Medical Research in National Taiwan University Hospital.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Chiu, Wc., Juang, Jm., Chang, Sn. et al. Angiotensin II regulates the LARG/RhoA/MYPT1 axis in rat vascular smooth muscle in vitro. Acta Pharmacol Sin 33, 1502–1510 (2012). https://doi.org/10.1038/aps.2012.117
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2012.117
Keywords
This article is cited by
-
Apatinib Through Activating the RhoA/ROCK Signaling Pathway to Cause Dysfunction of Vascular Smooth Muscle Cells
Applied Biochemistry and Biotechnology (2022)
-
DL0805-2, a novel indazole derivative, relaxes angiotensin II-induced contractions of rat aortic rings by inhibiting Rho kinase and calcium fluxes
Acta Pharmacologica Sinica (2016)
