
Abstract
Aim:
To investigate the mechanism underlying the increase of hepatoma cell proliferation by hepatitis B virus X protein (HBx).
Methods:
HepG2, H7402 and HepG2.2.15 cells, which constitutively replicated hepatitis B virus were used. The effects of HBx on hepatoma cell proliferation were examined using 5-ethynyl-2-deoxyuridine (EdU) incorporation assay and MTT assay. The expression level of MEKK2 was measured using RT-PCR, Western blot and luciferase reporter gene assay. The activity of activator protein 1 (AP-1) was detected using luciferase reporter gene assay. The phosphorylation levels of JNK and c-Jun were measured using Western blot. The expression levels of HBx and MEKK2 in 11 clinical hepatocellular carcinoma (HCC) tissues were measured using real time PCR and Western blot. In addition, the expression of MEKK2 in 95 clinical HCC tissues was examined using immunohistochemistry.
Results:
HBx significantly enhanced HepG2-X cell proliferation. In HepG2-X, H7402-X and HepG2.2.15 cells, the expression level of MEKK2 was remarkably increased. In HepG2.2.15 cells, HBx was found to activate JNK and AP-1, which were the downstream effectors of MEKK2 in HepG2-X and HepG2.2.15 cells. In 11 clinical HCC tissues, both HBx and MEKK2 expression levels were remarkably increased, as compared to those in the corresponding peritumor tissues. In 95 clinical HCC tissues, the rate of detection of MEKK2 was 85.3%.
Conclusion:
HBx promotes hepatoma cell proliferation via upregulating MEKK2, which may be involved in hepatocarcinogenesis.
Similar content being viewed by others


Introduction
Infection with the hepatitis B virus (HBV) is a major cause of acute hepatitis, fulminant hepatitis, and chronic hepatitis. Chronic infection is closely associated with the development of liver cirrhosis and hepatocellular carcinoma (HCC)1, 2. The HBV X protein (HBx) is involved in the pathogenesis of HBV-associated liver diseases by affecting gene expression, apoptosis, cell cycle and cell proliferation3, 4, 5. The expression of HBx induced the transformation of human liver cells and caused the development of hepatocellular carcinoma in certain strains of transgenic mice6, 7. In addition, HBx is involved in many cell signaling transduction pathways, such as the mitogen-activated protein kinase (MAPK), c-jun N-terminal kinase (JNK), phosphatidylinositol 3-kinase (PI 3-kinase), and JAK/STAT pathways8, 9, 10, 11. Our laboratory has focused on the investigation of HBx-mediated hepatocarcinogenesis. Our findings show that arachidonic acid metabolism, extracellular signal-related kinases (ERK1/2), sterol regulatory element binding protein 1c, 5-lipoxygenase and fatty acid synthase are involved in HBx-induced cell proliferation5, 12, 13. However, the underlying mechanisms that mediate the effects of HBx on cell proliferation require further investigation.
MAPK family members consist of at least four groups, including ERK1/2, JNK1/2/3, p38 proteins (p38a/b/c/d), and ERK3/5/714. In response to different external stimuli, these MAPKs are activated via a module that uses a MAPKKK-MAPKK-MAPK pathway. Mitogen-activated protein kinase kinase kinase 2 (MEKK2), which is a member of the MAPK signaling pathway, activates JNK and ERK515, 16. MEKK2 knockout mouse studies have identified important functions for MEKK2 in the T-cell receptor, epidermal growth factor (EGF) and fibroblast growth factor 2 (FGF-2) signaling pathways15, 17, 18. MEKK2 is involved in osteoblast activity, bone homeostasis and rheumatoid arthritis19, 20. HBx activates AP-1 through ERK and JNK does so through MEKK121. Although MEKK2 and MEKK1 are from the same mitogen-activated protein kinase kinase kinase (MAP3K) family, MEKK2, and MEKK1 are involved in different signaling pathways. For example, growth factor-induced urokinase-type plasminogen activator (uPA) expression requires MEKK1 and not MEKK222. Conversely, MEKK2, but not MEKK1, is required for mast cell tyrosine kinase receptor signaling23. Moreover, a recent study orthogonally confirmed the ability of MEKK2 to discriminate tumor cells from normal cells24. However, there are no data investigating the role of MEKK2 in HBx-mediated hepatocarcinogenesis. Therefore, we are interested in the role of MEKK2 in HBx-induced growth of hepatoma cells.
In the current study, we investigated the mechanism of increased cell proliferation, which is promoted by HBx. Our results show that HBx enhances the growth of hepatoma cells through upregulation of MEKK2. Our findings provide new insights into the mechanism that is employed by HBx to promote the proliferation of hepatoma cells.
Materials and methods
Cell culture
The cells of HepG2, H7402, and HepG2.2.15, which constitutively replicated HBV12, were maintained in Dulbecco's modified Eagle's medium (Life Technologies, Inc, Gaithersburg, MD, USA) supplemented with 10% fetal calf serum (Life Technologies). Cultures were incubated at 37 °C in a humidified atmosphere with 5% CO2. The engineered cells of HepG2-X/H7402-X (stably transfected with the pCMV-HBx plasmid), and HepG2-P/H7402-P (stably transfected with the empty pcDNA3.0 vector plasmid) were generated in HepG2 and H7402 cells, respectively, using the Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol, as previously described12.
Cell proliferation assay
HepG2 cells were seeded onto 96 well plates (1000 cells/well) for 24 h before transfection. MTT assays were used to assess cell proliferation every day from the first day until the fifth day after transfection. The MTT assay was performed as follows: 20 μL of MTT (5 mg/mL) was added to each well and the plates were incubated at 37 °C for 4 h. The MTT medium mixture was then removed and 150 μL of dimethyl sulfoxide (DMSO) was added to each well. The absorbance was measured at 570 nm using a multiwell spectrophotometer. The 5-ethynyl-2-deoxyuridine (EdU) incorporation assay was performed using the Cell-Light EdU imaging detecting kit according to the manufacturer's instructions (RiboBio, Guangzhou, China). EdU is a thymidine analog that is used in a similar manner as BrdU to label cells undergoing DNA replication25.
RNA extraction, reverse transcription PCR and real time PCR analysis
Total cellular RNA was extracted using the Trizol reagent (Invitrogen) from cells 48 h after treatment. cDNA was synthesized using the M-MLV RTase cDNA Synthesis Kit (TaKaRa Bio, Dalian, China) according to the manufacturer's protocol. To examine the expression of MEKK2 and HBx, we used specific primers for each gene as follows: MEKK2 gene (forward primer, 5′-TTTCCTCAAACGGATTT-3′ and reverse primer, 5′-TGTCTTCCCATCGTCA-3′) and HBx gene (forward primer, 5′-ATGGCTGCTAGGGTGTGCTG-3′ and reverse primer, 5′-CTAGGCAGAGGTGAAAAAGTTGC-3′). As a control, GAPDH was amplified using specific primers (forward, 5′-CATCACCATCTTCCAGGAGCG-3′ and reverse, 5′-TGACCTTGCCCACAGCCTTG-3′). Real-time PCR was performed using double-stranded DNA specific SYBR Green (TaKaRa Bio) and a Bio-Rad sequence detection system according to the manufacturer's instructions. Relative expression was determined by the comparative Ct method using 2-ΔΔ Ct26.
Western blot analysis
Cells were washed in PBS, and cellular proteins were extracted in RIPA buffer (Biomed, Beijing, China) for 30 min at 4 °C. Lysates were cleared by centrifugation, and proteins were separated by gel electrophoresis. For tissue protein extraction, frozen tissues were quickly ground into a fine powder using a mortar and pestle under liquid nitrogen and prepared for Western blot analysis as described above. Membranes were blocked in PBS containing 0.1% Tween20 (PBS-T) and 5% (w/v) milk for 1 h at room temperature. Membranes were then incubated with primary antibodies diluted in PBS-T for 2 h at room temperature. Subsequently, membranes were washed with PBS-T and incubated with peroxidase-conjugated secondary antibody that was diluted in PBS-T at room temperature for 1 h. Membranes were washed in PBS-T and bound antibody was detected using the enhanced chemiluminescence system ECL-plus Reagents (Amersham Biosciences, Buckinghamshire, UK). After 48 h of transfection, Western blot analysis was performed as described above. The primary antibodies were mouse anti-MEKK2 (Santa Cruz, CA, USA), anti-HBx (Abcam, Cambridge, UK), anti-diphosphorylated JNK (Sigma, St Louis, MO, USA), anti-JNK (Sigma), anti-phosphorylated c-Jun (Cell signaling, Danvers, MA, USA) and mouse anti-β-actin (Sigma).
RNA interference
pSilencer-HBx (pSi-HBx) was used to produce small interfering RNAs (siRNAs) to target HBx mRNA, and pSilencer-control (pSi-con) was used as a negative control27, 28. siRNA duplexes targeting human MEKK2 were synthesized and purified by RiboBio (Guangzhou, China)29. siRNA duplexes containing non-specific sequences were used as a negative control (NC). Different siRNAs were transfected separately into cells using the Lipofectamine 2000 (Invitrogen) reagent, and the medium was replaced 6 h after transfection.
Construction of the human MEKK2 promoter
To generate a clone of the MEKK2 promoter in a pGL3 basic vector (Promega, Madison, WI, USA), a ∼1800-bp region of the putative promoter (from +83 to -1814 nt) was amplified from HepG2 genomic DNA by PCR using specific primers (forward primer, 5′-CCGCTCGAGCTGTAGTCCCATCTACTCGG-3′ and reverse primer, 5′-CCCAAGCTTTTAGTTTGAACTCACCCATT-3′ and was cloned into the pGL3-basic vector.
Luciferase reporter gene assay
Transfected cells were harvested after 36 h. Luciferase activity was determined using the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer's instructions. The firefly luciferase activity was normalized to Renilla luciferase expression for each sample. All experiments were performed at least three times. pAP1-Luc (Clontech, Plano Alto, CA, USA) is a firefly luciferase reporter gene vector that contains AP-1 binding sites.
Patient samples
The 11 clinical HBV-positive HCC tissues and their corresponding nearby noncancerous livers that were used in this study were obtained from patients who underwent radical resection at Tianjin First Center Hospital (Tianjin, China). All of the tumor samples were immediately frozen after surgical resection and stored in liquid nitrogen until analysis. Written consent, which approved the use of their tissues for research purposes after the operation, was obtained from the patients. The study was approved by the Institute Research Ethics Committee at the Nankai University.
Immunohistochemistry
The hepatocellular carcinoma tissue microarrays were obtained from the Xi-an Aomei Biotechnology Co, Ltd (Xi-an, China). These microarrays were composed of 95 hepatocellular carcinoma (HCC) tissue samples (18 females, 77 males, average age 61 years), which included duplicate core biopsies (1 mm in diameter) from fixed, paraffin-embedded tumors. The immunohistochemical staining of samples was performed as previously reported27. The primary antibody of mouse anti-MEKK2 was used (1:50, Abcam). Immunostained slides were evaluated under the microscope. The percentage of cells showing positive nuclear and/or cytoplasmic staining for MEKK2 was calculated by reviewing the entire slide. Based on the percentage of cells with positive nuclear and/or cytoplasmic staining, staining patterns were classified using a six-grade scale as follows: 0, the absence of cell nuclear and/or cytoplasmic staining; 1+, <10% cells with positive nuclear and/or cytoplasmic staining; 2+, 10%–25% positive cells; 3+, 26%–50% positive cells; 4+, 51%–75% positive cells; 5+, >75% positive cells. For statistical analysis, HCC patients were subgrouped into MEKK2-negative staining (scale 0) and MEKK2 positive-staining (scale 1–5) groups. The categorization of immunostaining intensity was performed by three independent observers. The negative control, which lacked incubation with the primary antibody, was treated as described above.
Statistical analysis
All data were expressed as the mean±SD. Statistical analysis was performed using student's t test. P<0.05 was indicated to be statistically significant.
Results
HBx enhances the proliferation of hepatoma cells
Previously, we established two engineered cell lines, namely HepG2-X, which was stably transfected with HBx, and HepG2-P, which was stably transfected with an empty pcDNA3.0 vector13. We examined the growth of HepG2-X cells using the EdU incorporation assay and MTT assay. The results showed that HepG2-X cells grew faster than HepG2-P or HepG2 cells (Figures 1A and 1B). In addition, RNA interference (RNAi) targeting HBx mRNA abolished HBx-induced growth of HepG2 cells. Therefore, the results confirm that HBx enhances the proliferation of hepatoma cells.

HBx enhances the proliferation of HepG2-X cells. (A, B) The proliferation ability of hepatoma cells was tested using the 5-ethynyl-2-deoxyuridine (EdU) incorporation and MTT assays. Results are representative of three independent experiments. Values represent mean±SD. bP<0.05, cP<0.01, student's t test.
HBx upregulates the expression of MEKK2
Previously, we showed using cDNA microarrays that a broad range of genes show aberrant expression in hepatoma cells that overexpress HBx, the results of which included MEKK2 upregulation30. To investigate whether MEKK2 is involved in the HBx-mediated growth of hepatoma cells, we further confirmed the effect of HBx on expression of MEKK2. The results show that MEKK2 is significantly upregulated in HepG2-X (or H7402-X) cells at the levels of mRNA and protein, as assessed by RT-PCR and immunoblot analysis (Figure 2A). Meanwhile, we examined the changes of MEKK2 expression after RNA-mediated knockdown of the HBx expression. As expected, the expression level of MEKK2 was decreased in a dose-dependent manner (Figure 2A). Moreover, Western blot analysis showed that MEKK2 was upregulated in HepG2.2.15 cells compared with HepG2 cells (Figure 2B). To investigate the effect of HBx on the promoter activity of MEKK2, we generated a MEKK2 promoter luciferase reporter, which was termed pGL3-MEKK2 pro. Additionally, transient transfection was performed in HepG2 and H7402 cells using the pGL3-MEKK2 promoter plasmid. The promoter activity of MEKK2 was examined in HepG2/H7402 cell lines using luciferase reporter gene assays. The data confirmed the successful construction of the plasmid (Figure 2C). The promoter activity of MEKK2 was remarkably enhanced in HepG2-X/H7402-X cells relative to that in HepG2/H7402 cells (Figure 2D). Meanwhile, the luciferase reporter gene assay showed that HBx RNAi abolished the enhancement of MEKK2 promoter activity in HepG2-X/H7402-X/HepG2.2.15 cells in a dose-dependent manner (Figure 2E).

HBx upregulates the expression of MEKK2. (A) The levels of MEKK2 and HBx were detected using RT-PCR and immunoblot analysis. GAPDH and β-actin were used as internal controls. (B) The levels of MEKK2 and HBx were detected in HepG2.2.15 cells using immunoblot analysis. (C) The plasmids of pGL3-MEKK2 pro and pGL3 basic vector were transfected into HepG2 and H7402 cells, respectively. The relative luciferase activity was tested using the luciferase reporter gene assay. (D) The promoter activities of MEKK2 were examined in HepG2-X/H7402-X cells. (E) The promoter activities of MEKK2 were measured after treatment with RNAi that targeted HBx mRNA in HepG2-X/H7402-X/HepG2.2.15 cells in a dose-dependent manner. Results are representative of three independent experiments. Values represent mean±SD. bP<0.05, cP<0.01, student's t test.
It has been reported that MEKK2 is involved in the activation of AP-1 and JNK15, 16, 19. To further confirm the HBx-mediated regulation of MEKK2, we examined the downstream effectors of MEKK2, such as AP-1 and JNK, in HepG2.2.15 cells. The luciferase report gene assay showed that the activity of AP-1 was decreased in the cells that were treated with HBx RNAi (Figure 3A). AP-1 activation is induced in part by the phosphorylation of c-Jun31. To further confirm the luciferase report gene assay result, the phosphorylation level of c-Jun was examined. The Western blot analysis showed that the protein level of MEKK2 and the phosphorylation levels of JNK and c-Jun were decreased (Figure 3B). We silenced the expression of MEKK2 in HepG2.2.15 cells. The data revealed that the knockdown of MEKK2 reduced the activity of AP-1 (Figure 3C). Meanwhile, the phosphorylation levels of JNK and c-Jun were decreased (Figure 3D). Therefore, we conclude that HBx upregulates MEKK2.

HBx upregulates the expression of MEKK2 to mediate AP-1 and JNK activation. (A) The activity of AP-1 was determined in HepG2.2.15 cells using the luciferase reporter gene assay after HBx RNAi treatment. (B) The phosphorylation levels of JNK and c-Jun were determined using immunoblot analysis. (C) The activity of AP-1 was determined in HepG2.2.15 cells using the luciferase reporter gene assay after MEKK2 RNAi treatment. (D) The phosphorylation levels of JNK and c-Jun were determined using immunoblot analysis. The results are representative of three independent experiments. Values represent mean±SD. cP<0.01, student's t test.
MEKK2 contributes to HBx-mediated growth of hepatoma cells
The function of MEKK2 in hepatoma cells is unclear. To elucidate the function of MEKK2, we silenced the expression of MEKK2 in HepG2-X cells using specific siRNAs. The cell proliferation was then measured using EdU incorporation assay and MTT assay. Our results demonstrate that the growth of HepG2-X cell was inhibited by MEKK2 siRNAs (Figure 4A and 4B). Similar effects were observed in HepG2.2.15 cells (Figure 4C and 4D). These results suggest that MEKK2 is involved in HBx-mediated growth of hepatoma cells.

MEKK2 contributes to HBx-mediated growth of hepatoma cells. (A, B) The proliferation ability of HepG2-X cells was tested using the EdU incorporation and MTT assays after MEKK2 siRNA treatment. (C, D) The proliferation ability of HepG2.2.15 cells was tested using the EdU incorporation and MTT assays after MEKK2 siRNA treatment. Results are representative of three independent experiments. Values represent mean±SD. bP<0.05, cP<0.01 vs NC, student's t test.
MEKK2 is overexpressed in clinical HCC tissues
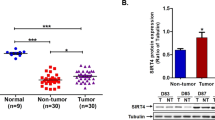
We examined the expression level of MEKK2 in 11 cases of human liver cancer tissues with HBV infection at the mRNA and protein levels using real time PCR and Western blot analysis. Real time PCR analysis revealed that all 11 clinical HCC tissues and their respective peritumor tissues were positive for HBx expression. The expression level of HBx was higher in HCC tissues relative to that in their respective peritumor tissues (Figure 5A). The expression level of MEKK2 was also higher in tumor tissues relative to their peritumor tissues (Figure 5B). Consistent with the results from real time PCR, Western blot analysis revealed that HBx was upregulated in 8 of 11 cases of HCC tissues, whereas MEKK2 was upregulated in 7 of 11 cases (Figure 5C). The clinical data strongly support the conclusion that MEKK2 expression is positively correlated with HBx expression in carcinogenesis. The expression of MEKK2 was assessed using immunohistochemical staining on a tissue microarray that contained 95 cases of HCC tissues. The data showed that the percentage of MEKK2 positivity was 85.3% (81/95 cases) in HCC tissues (Figure 5D).

MEKK2 is overexpressed in clinical HCC tissues. (A, B) The relative mRNA expression levels of HBx and MEKK2 were examined in clinical HBx-positive HCC tissues using real time PCR. Values represent mean±SD. bP<0.05, cP<0.01, Student's t test. (C) The expression levels of HBx and MEKK2 were examined in HCC patients using Western blot analysis. Protein bands were quantified using Quantity One software (Bio-Rad). The value under each pair of samples (T/P), represents the ratio of HBx or MEKK2 expression in the HCC tissue to that in its corresponding peritumor tissue and indicates the fold change in the protein level in HCC. P, peritumor tissue; T, HCC tissue. (D) Immunohistochemical staining showed the expression levels of MEKK2 in HCC tissues using a tissue microarray. (a) Negative control; (b) MEKK2-positive staining. Imaged at 200× magnification.
Discussion
HBx is an essential viral protein that is involved in viral pathogenesis and carcinogenesis. HBx strongly enhances the growth of hepatoma cells in a system of stable transfection5, 12, 13, 32. In the current study, we further investigated the mechanism that HBx employs to enhance the proliferation of hepatoma cells. Previously, we examined the expression profiles in stably HBx-transfected hepatoma H7402 (H7402-X) cells using cDNA microarrays30. We determined that MEKK2 was one of the upregulated genes. Therefore, we further investigated the role of MEKK2 in HBx-mediated hepatocarcinogenesis.
Previous studies have shown that HBx exhibits either apoptotic or proliferative activities in a context-dependent manner33, 34. HBx promotes proliferation in stably transfected cells5, 12, 13, 32. Therefore, we investigated the HBx-mediated proliferation in a model of stably HBx-transfected hepatoma HepG2-X cells. We confirmed our previous findings that HBx enhances the proliferation of hepatoma cells in our system (Figure 1). We demonstrated that HBx upregulated MEKK2 at the mRNA and protein level (Figure 2A and 2B). The transcriptional regulation that is mediated by the promoter is a key event in the regulation of gene expression. We demonstrated that HBx activated the promoter activity of MEKK2 in hepatoma cells (Figure 2D). HBx activates AP-1 via a pathway that is mediated by the activation of ERK and JNK21, 35. AP-1 and JNK are downstream effectors of MEKK215, 16, 19. To further validate that HBx upregulated MEKK2, we examined the activation of AP-1 and JNK by silencing the expression of HBx or MEKK2 in HepG2.2.15 cells. Our results demonstrate that activation of AP-1 and JNK was inhibited in the cells after siRNA treatment (Figure 3) and support the conclusion that HBx upregulates MEKK2.
At present, few studies have investigated MEKK2 function, even though MEKK2 is involved in osteoblast activity, bone homeostasis and rheumatoid arthritis19, 20. In the current study, we demonstrated the function of MEKK2 in hepatoma cells. Our results show that MEKK2 promotes the growth of hepatoma HepG2-X cells and HepG2.2.15 cells, which was characterized using the EdU incorporation and MTT assays (Figure 4). These results suggest that MEKK2 is involved in HBx-mediated proliferation in hepatoma cells. Previously, the clinical significance of MEKK2 in hepatoma tissues has not been elucidated. Therefore, we examined the levels of MEKK2 in 11 cases of clinical HBx-positive hepatoma tissues using real time PCR and Western blot analysis (Figure 5A). Interestingly, we found that the levels of HBx and MEKK2 expression were higher in tumor tissues compared to that in peritumor tissues (Figure 5B). These results suggest that the expression of MEKK2 is positively correlated with HBx expression in carcinogenesis. We are the first to report that the percentage of MEKK2-positive staining is very high (85.3%) in HCC tissues (Figure 5D). This result suggests that MEKK2 is potentially involved in hepatocarcinogenesis.
In summary, we conclude that HBx upregulates the expression of MEKK2 and that MEKK2 is involved in the promotion of hepatoma cell proliferation, which is mediated by HBx. Our finding suggests that MEKK2 may play important roles in the hepatocarcinogenesis mediated by HBx.
Author contribution
Xiao-dong ZHANG, Guang-yao KONG, and Li-hong YE designed the research; Guang-yao KONG, Jun-ping ZHANG, Shuai ZHANG, and Chang-liang SHAN performed the research; Xiao-dong ZHANG, Guang-yao KONG, and Li-hong YE analyzed the data and wrote the paper.
References
Koike K . Hepatitis B virus X gene is implicated in liver carcinogenesis. Cancer Lett 2009; 286: 60–8.
Zhang X, Zhang H, Ye L . Effects of hepatitis B virus X protein on the development of liver cancer. J Lab Clin Med 2006; 147: 58–66.
Liang X, Liu Y, Zhang Q, Gao L, Han L, Ma C, et al. Hepatitis B virus sensitizes hepatocytes to TRAIL-induced apoptosis through Bax. J Immunol 2007; 178: 503–10.
Mukherji A, Janbandhu VC, Kumar V . HBx-dependent cell cycle deregulation involves interaction with cyclin E/A-cdk2 complex and destabilization of p27Kip1. Biochem J 2007; 401: 247–56.
Shan C, Xu F, Zhang S, You J, You X, Qiu L, et al. Hepatitis B virus X protein promotes liver cell proliferation via a positive cascade loop involving arachidonic acid metabolism and p-ERK1/2. Cell Res 2010; 20: 563–75.
Zhang WY, Cai N, Ye LH, Zhang XD . Transformation of human liver L-O2 cells mediated by stable HBx transfection. Acta Pharmacol Sin 2009; 30: 1153–61.
Kim CM, Koike K, Saito I, Miyamura T, Jay G . HBx gene of hepatitis B virus induces liver cancer in transgenic mice. Nature 1991; 351: 317–20.
Kekule AS, Lauer U, Weiss L, Luber B, Hofschneider PH . Hepatitis B virus transactivator HBx uses a tumour promoter signalling pathway. Nature 1993; 361: 742–5.
Natoli G, Avantaggiati ML, Chirillo P, De Marzio E, Collepardo D, Falco M, et al. Modulation of intracellular signal transduction pathways by the hepatitis B virus transactivator pX. J Hepatol 1995; 22: 14–20.
Lee YH, Yun Y . HBx protein of hepatitis B virus activates Jak1-STAT signaling. J Biol Chem 1998; 273: 25510–5.
Diao J, Khine AA, Sarangi F, Hsu E, Iorio C, Tibbles LA, et al. X protein of hepatitis B virus inhibits Fas-mediated apoptosis and is associated with up-regulation of the SAPK/JNK pathway. J Biol Chem 2001; 276: 8328–40.
Wang Q, Zhang WY, Ye LH, Zhang XD . A mutant of HBx (HBxDelta127) promotes hepatoma cell growth via sterol regulatory element binding protein 1c involving 5-lipoxygenase. Acta Pharmacol Sin 2010; 31: 367–74.
Wang Q, Zhang W, Liu Q, Zhang X, Lv N, Ye L, et al. A mutant of hepatitis B virus X protein (HBxDelta127) promotes cell growth through a positive feedback loop involving 5-lipoxygenase and fatty acid synthase. Neoplasia 2010; 12: 103–15.
Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, et al. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev 2001; 22: 153–83.
Su B, Cheng J, Yang J, Guo Z . MEKK2 is required for T-cell receptor signals in JNK activation and interleukin-2 gene expression. J Biol Chem 2001; 276: 14784–90.
Chayama K, Papst PJ, Garrington TP, Pratt JC, Ishizuka T, Webb S, et al. Role of MEKK2-MEK5 in the regulation of TNF-alpha gene expression and MEKK2-MKK7 in the activation of c-Jun N-terminal kinase in mast cells. Proc Natl Acad Sci U S A 2001; 98: 4599–604.
Schaefer BC, Ware MF, Marrack P, Fanger GR, Kappler JW, Johnson GL, et al. Live cell fluorescence imaging of T cell MEKK2: redistribution and activation in response to antigen stimulation of the T cell receptor. Immunity 1999; 11: 411–21.
Sun W, Wei X, Kesavan K, Garrington TP, Fan R, Mei J, et al. MEK kinase 2 and the adaptor protein Lad regulate extracellular signal-regulated kinase 5 activation by epidermal growth factor via Src. Mol Cell Biol 2003; 23: 2298–308.
Yamashita M, Ying SX, Zhang GM, Li C, Cheng SY, Deng CX, et al. Ubiquitin ligase Smurf1 controls osteoblast activity and bone homeostasis by targeting MEKK2 for degradation. Cell 2005; 121: 101–13.
Hammaker DR, Boyle DL, Chabaud-Riou M, Firestein GS . Regulation of c-Jun N-terminal kinase by MEKK-2 and mitogen-activated protein kinase kinase kinases in rheumatoid arthritis. J Immunol 2004; 172: 1612–8.
Jacqueline B, Fei S, Margherita D, Robert JS . Hepatitis B virus HBx protein induces transcription factor AP-1 by activation of extracellular signal-regulated and c-Jun N-terminal mitogen-activated protein kinases. J Virol 1996; 70: 4978–85.
Witowsky J, Abell A, Johnson NL, Johnson GL, Cuevas BD . MEKK1 is required for inducible urokinase-type plasminogen activator expression. J Biol Chem 2003; 278: 5941–6.
Garrington TP, Ishizuka T, Papst PJ, Chayama K, Webb S, Yujiri T, et al. MEKK2 gene disruption causes loss of cytokine production in response to IgE and c-Kit ligand stimulation of ES cell-derived mast cells. EMBO J 2000; 19: 5387–95.
Cazares LH, Troyer D, Mendrinos S, Lance RA, Nyalwidhe JO, Beydoun HA, et al. Imaging mass spectrometry of a specific fragment of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase kinase 2 discriminates cancer from uninvolved prostate tissue. Clin Cancer Res 2009; 15: 5541–51.
Salic A, Mitchison TJ . A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc Natl Acad Sci U S A 2008; 105: 2415–20.
Wang FZ, Sha L, Zhang WY, Wu LY, Qiao L, Li N, et al. Involvement of hepatitis B X-interacting protein (HBXIP) in proliferation regulation of cells. Acta Pharmacol Sin 2007; 28: 431–8.
Zhang X, Dong N, Yin L, Cai N, Ma H, You J, et al. Hepatitis B virus X protein upregulates survivin expression in hepatoma tissues. J Med Virol 2005; 77: 374–81.
Zhang H, Shan CL, Li N, Zhang X, Zhang XZ, Xu FQ, et al. Identification of a natural mutant of HBV X protein truncated 27 amino acids at the COOH terminal and its effect on liver cell proliferation. Acta Pharmacol Sin 2008; 29: 473–80.
Winsauer G, Resch U, Hofer-Warbinek R, Schichl YM, de Martin R . XIAP regulates bi-phasic NF-kappaB induction involving physical interaction and ubiquitination of MEKK2. Cell Signal 2008; 20: 2107–12.
Ye L, Dong N, Wang Q, Xu Z, Cai N, Wang H, et al. Progressive changes in hepatoma cells stably transfected with hepatitis B virus X gene. Intervirology 2008; 51: 50–8.
Shaulian E, Karin M . AP-1 as a regulator of cell life and death. Nat Cell Biol 2002; 4: E131–6.
Wang F, Zhou H, Xia X, Sun Q, Wang Y, Cheng B . Activated Notch signaling is required for hepatitis B virus X protein to promote proliferation and survival of human hepatic cells. Cancer Lett 2010; 298: 64–73.
Kim KH, Seong BL . Pro-apoptotic function of HBV X protein is mediated by interaction with c-FLIP and enhancement of death-inducing signal. EMBO J 2003; 22: 2104–16.
Shih WL, Kuo ML, Chuang SE, Cheng AL, Doong SL . Hepatitis B virus X protein inhibits transforming growth factor-beta-induced apoptosis through the activation of phosphatidylinositol 3-kinase pathway. J Biol Chem 2000; 275: 25858–64.
Nijhara R, Jana SS, Goswami SK, Rana A, Majumdar SS, Kumar V, et al. Sustained activation of mitogen-activated protein kinases and activator protein 1 by the hepatitis B virus X protein in mouse hepatocytes in vivo. J Virol 2001; 75: 10348–58.
Acknowledgements
This work was supported by grants from the National Basic Research Program of China (973 Program, No 2007CB914802, 2007CB914804 and 2009CB521702) and the National Natural Science Foundation of China (81071624).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Kong, Gy., Zhang, Jp., Zhang, S. et al. Hepatitis B virus X protein promotes hepatoma cell proliferation via upregulation of MEKK2. Acta Pharmacol Sin 32, 1173–1180 (2011). https://doi.org/10.1038/aps.2011.52
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2011.52
Keywords
This article is cited by
-
The LPI/GPR55 axis enhances human breast cancer cell migration via HBXIP and p-MLC signaling
Acta Pharmacologica Sinica (2018)
-
MiR-19a, miR-122 and miR-223 are differentially regulated by hepatitis B virus X protein and involve in cell proliferation in hepatoma cells
Journal of Translational Medicine (2016)
-
Hepatitis B virus X protein promotes human hepatoma cell growth via upregulation of transcription factor AP2α and sphingosine kinase 1
Acta Pharmacologica Sinica (2015)
-
Hepatitis B virus X protein upregulates Lin28A/Lin28B through Sp-1/c-Myc to enhance the proliferation of hepatoma cells
Oncogene (2014)
