
Abstract
The G-protein-coupled receptors (GPCRs) are one of the largest super families of cell-surface receptors and play crucial roles in virtually every organ system. One particular family of GPCRs, the class C GPCRs, is distinguished by a characteristically large extracellular domain and constitutive dimerization. The structure and activation mechanism of this family result in potentially unique ligand recognition sites, thereby offering a variety of possibilities by which receptor activity might be modulated using novel compounds. In the present article, we aim to provide an overview of the exact sites and structural features involved in ligand recognition of the class C GPCRs. Furthermore, we demonstrate the precise steps that occur during the receptor activation process, which underlie the possibilities by which receptor function may be altered by different approaches. Finally, we use four typical family members to illustrate orthosteric and allosteric sites with representative ligands and their corresponding therapeutic potential.
Similar content being viewed by others


Introduction
The G-protein-coupled receptors (GPCRs) form the largest class of cell surface receptors and play a major role in cellular perception of the environment1. GPCRs are sensitive to a diverse range of ligands that include light (photons), ions, amino acids and large proteins, and they represent an important market for pharmaceutical companies. Approximately 50 GPCRs are estimated to be targeted by nearly half of the currently marketed drugs, and at least 300 GPCRs remain to be exploited2. Intense efforts have been devoted to screening new GPCR ligands that display high potential as drug leads. However, for many GPCRs, such efforts have failed to yield viable drug candidates. Numerous issues prohibit traditional GPCR-targeted drug discovery. For instance, ligands screened by traditional techniques usually act on GPCR orthosteric sites. The conserved characteristics of the orthosteric sites make it difficult to achieve high selectivity for specific GPCR subtypes. Furthermore, the persistent treatment regime of orthosteric ligands often leads to potent side effects and tolerance to the drugs. In addition, for some GPCRs, such as peptide or protein receptors, it is inherently difficult to design synthetic orthosteric ligands. Therefore, the pharmaceutical industry is searching for alternative approaches to identify new modulators of GPCRs. The determination of GPCR structures, mechanisms and ways in which to modulate these properties are therefore of critical importance.
The GPCRs can be classified into five families based on the sequence phylogeny of a conserved heptahelical transmembrane domain (7TM)3. Among these families, class C GPCRs are defined by two unique structural features: first, they possess a large extracellular domain that is distal to the 7TM and contains the orthosteric sites; second, they form constitutive dimers with unique activation modes compared with other classes of GPCRs4. Class C GPCRs are composed of metabotropic glutamate receptors (mGlu receptors), γ-aminobutyric acidB receptors (GABAB receptors), Ca2+-sensing receptors (CaS receptors), sweet and amino acid taste receptors, pheromone receptors, odorant receptors in fish and several orphan receptors3. mGlu, GABAB, and CaS receptors represent an important new class of therapeutic targets that are integral to disorders that affect the central neural system (CNS) and calcium homeostasis4, 5. The taste receptors, on the other hand, attract significant attention from food companies because the taste additives that target these receptors represent a key feature of the large food industry market5.
The recently identified class C GPCRs have been targeted by only two therapeutic drugs currently on the market6. By contrast, in recent years there have been tremendous advances in the discovery of allosteric modulators of class C GPCRs, most likely as a result of the existence of multiple modulation sites for various ligands7. Cinacalcet, one of the first two allosteric modulators of GPCRs on the market, targets the CaS receptor5. This review focuses on the structural features that are involved in ligand recognition by class C GPCRs. The possibilities of modulating receptor function through different types of ligands are then discussed. Finally, representative ligands and the associated sites of four typical family members that contain therapeutic potential are reviewed in detail. The ligands described in this review are small chemical molecules. Peptide ligands, such as antibodies, are not discussed.
Representative family members
L-Glutamate serves as the neurotransmitter at the majority of excitatory synapses in the mammalian CNS. As the metabotropic receptors for glutamate, mGlu receptors participate in the modulation of synaptic transmission and neuronal excitability throughout the CNS8, 9. The mGlu receptors are sub-classified into three groups based on sequence homology, G-protein coupling, and ligand selectivity9. Group I (mGlu1 and 5) couple to Gq/G11 and activate phospholipase Cβ, resulting in the hydrolysis of phosphoinositides and the generation of inositol 1,4,5-trisphosphate (IP3) and diacylglycerol, whereas Group II (mGlu 2 and 3) and Group III (mGlu 4, 6, 7, and 8) couple predominantly to Gi/o, which inhibits adenylyl cyclase and directly regulates ion channels and other downstream signaling partners via the liberation of Gβγ subunits10. The widespread expression of mGlu receptors makes these receptors particularly attractive drug targets, and recent studies continue to validate the therapeutic utility of mGlu receptor ligands in neurological and psychiatric disorders, such as Parkinson's disease11, Fragile X syndrome12, Alzheimer's disease13, anxiety, and schizophrenia14.
GABA is a major inhibitory neurotransmitter in the mammalian CNS. As the metabotropic receptor for GABA, GABAB receptor mediates slow and prolonged synaptic inhibition15. The GABABreceptor functions as an obligate heterodimer of two subtypes, GABAB1 and GABAB216, 17. GABAB1 contains the GABA binding site18, while GABAB2 is responsible for Gi/o protein activation19. In addition to a role in neuronal excitability and plasticity, GABAB receptor may promote neuron survival under conditions of metabolic stress20, ischemia21, or apoptosis22. This receptor is a promising target for the treatment of many diseases, including spasticity, neuropathic pain23, drug addiction, schizophrenia, anxiety, depression and epilepsy24, 25.
The CaS receptor is a unique class C GPCR that can be activated by ions without the cooperation of other ligands4. This receptor is highly sensitive to a very slight change in extracellular Ca2+ concentrations, which ensures its significant role in regulating calcium homeostasis26. The CaS receptor is involved in several disorders, including hyperparathyroidism, osteoporosis and different forms of hypocalcemia26, 27, 28. The clinical success of Cinacalcet indicates that more efforts should be devoted to the discovery of novel ligands that modulate CaS receptor activation.
The class C GPCRs contain three taste receptor subunits (T1R1, T1R2, and T1R3) that form two heterodimers, the sweet receptor (T1R2/T1R3) and the umami receptor (T1R1/T1R3)29, 30. Only cis activation occurs within the sweet and umami taste receptors, which means T1R2 in the sweet receptor or T1R1 in the umami receptor are involved in both orthosteric ligand recognition and in G protein activation, whereas the common subunit T1R3 loses the corresponding function31. In addition to natural sugars, the sweet taste receptor is also sensitive to artificial sweeteners, sweet proteins and some D-amino acids. In most mammals, the umami receptor can be activated by L-amino acids, whereas the human orthologue is only sensitive to monosodium glutamate and L-aspartate. Flavor enhancers, such as purine nucleotides, have the ability to potentiate umami receptor function. These artificial sweeteners and flavor enhancers represent a large food sector market6.
Structural features of class C GPCRs
Class C GPCRs are composed of an exceptionally large extracellular domain, a heptahelical transmembrane domain and an intracellular carboxyl-terminal (C-terminal) domain (Figure 1A). One distinct structural feature of class C GPCRs is the extracellular domain that contains a Venus flytrap (VFT) module and a cysteine rich domain (CRD, except in the GABAB receptor). The 7TM domain is conserved among all GPCRs with the exception that class C GPCR 7TMs contain only the allosteric sites. The orthosteric sites are contained within the VFT. The C-terminal tail of class C GPCRs is a highly variable domain and plays a role in scaffolding and signaling protein coupling3. All the domains except for the intracellular C-terminal domain provide plentiful ligand action sites. The other unique characteristic of class C GPCRs is their mandatory dimerization, either as homodimers (mGlu and CaS receptors) or heterodimers (GABAB receptor and T1Rs) (Figure 1B). The allosteric interaction between different dimer domains results in a particularly complicated activation process.

Schematic structure of class C GPCRs. (A) Structural organization of class C GPCRs. Class C GPCRs have a common structure consisting of a VFT with two lobes (lobe 1 and lobe 2) separating by a cleft as orthosteric site, a 7TM and a CRD for all but GABAB receptor. The crystal structure of mGlu3 receptor (PDB ID 2E4W) was used for the VFT and CRD. The bovine rhodopsin crystal structure (PDB ID 1GZM) was used for the 7TM. (B) Schematic representation of two prototypical class C GPCRs as heterodimer (GABAB receptor), or homodimer (mGlu receptor).For GABAB receptor, the VFT is directly linked to the 7TM. Two subunits, GABAB1 and GABAB2, form an obligatory heterodimer. GABAB1 is responsible for endogenous ligands binding, while GABAB2 is responsible for G protein activating. For mGlu receptors, the VFT connects to the 7TM via CRD. mGlu receptors form homodimers which could offer two orthosteric sites per dimer. (C) The determined cystal structure for the VFT and CRD. The first solved structure is the VFT of mGlu1 receptor (PDB ID 1EWK), which shows that the VFT oscillates between an open and a closed conformation. The crystal structure of whole extracellular domain including the VFT and CRD (PDB ID 2E4W) has been solved firstly in mGlu3 receptor.
Extracellular domain
Venus flytrap module
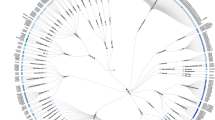
The VFTs of class C GPCRs share sequence and structural similarity with bacterial periplasmic binding proteins (PBPs)31. A generally accepted hypothesis is that the fusion of an ancestral rhodopsin-like receptor and a PBP formed the common ancestor of the class C GPCRs3. Additional detailed phylogenetic analysis of VFTs from four typical groups of class C GPCRs reveals that functional divergence involved positive selection and is partially responsible for the evolutionary patterns of the VFTs (Figure 2)32. The functionally divergent sites could represent potential drug targets that participate in ligand recognition.

The bootstrap tree of the prototypical members from human class C GPCRs. The sequence of the VFT were aligned using the default parameters and the homologous bacterial PBPs were used as an outgroup to root the trees. Class C GPCRs form obligatory dimers. Homodimers (mGluR and CaSR) linked by a disulfide bond between their VFTs, while heterodimers (GABABR and T1R) are not covalently linked.
Among class C GPCRs, the VFT of the mGlu1 receptor is the first for which a crystal structure was solved, both in the absence and presence of its orthosteric ligands (Figure 1C)33. The crystal structure of VFT revealed a bilobate domain with two lobes being separated by a cleft in which endogenous ligands bind33, 34. The VFT oscillates between an open and closed conformation in the absence of bound ligand. In the presence of ligand, glutamate interacts with lobe 1 in the open form of the VFT and then stabilizes a closed form through additional contacts with lobe 2. Competitive antagonists inhibit receptor activation by preventing VFT closure35, whereas locking the VFT in a closed conformation with an artificial disulfide bond results in a constitutively active receptor36.
VFTs form constitutive dimers. Based on the crystal structure and mutational analysis of mGlu1 VFTs, the hydrophobic interaction between lobe 1 of each monomer is the main driving force for VFT dimerization37, 38. Additionally, a disulfide bond linking the two VFTs was demonstrated to stabilize this dimer37, 39, 40. Similar to mGlu receptors, lobe 1 from each of the two hetero-subunits of the GABAB receptor, GABAB1 and GABAB2, mediates subunit interaction41, 42. The lobe 1 N-glycan, which is located at the interface of either GABAB1 or GABAB2, prevents receptor heterodimerization and cell surface trafficking42.
Cysteine rich domain (CRD)
For most class C GPCRs (except for the GABAB receptor), the VFT and 7TM are connected by the CRD. The CRD is a roughly 80 amino acid segment that contains nine completely conserved cysteines3. The crystal structure of the complete extracellular domain of the mGlu3 receptor was solved in 2007 (Figure 1C)43. Based on this structure, the CRD forms an independent domain with a length of 40 Å, which physically separates the VFT and the 7TM. The CRD plays an important role in receptor activation of the mGlu receptors, CaS receptors and sweet taste receptors with subunit T1R344, 45. In mGlu-like receptors, a conserved disulfide bridge between the VFT and the CRD is required for the allosteric interaction between the VFT and the 7TM. Mutation of this disulfide bond abolished agonist-induced activation of the mGlu receptors46.
Heptahelical transmembrane domain (7TM)
Similar to other GPCRs, class C GPCRs possess heptahelical transmembrane helices that are linked by three short intra- (iloops) and extracellular loops, which are always smaller than 30 residues. Despite the low primary sequence similarity, several similar 3D structural features of the 7TM exist between the class C GPCRs and the rhodopsin-like receptors, including the conserved disulfide bond that connects the top of TM3 and the second extracellular loop, the central position of TM3, the 8th helix following the 7TM that is related to G-protein coupling as well as several conserved residues3.
In contrast to rhodopsin-like GPCRs, the 7TM of class C GPCRs does not participate in ligand recognition or binding. However, this domain in class C GPCRs still contains a conserved binding pocket that corresponds to the orthosteric sites of rhodopsin-like GPCRs47. This binding pocket represents a site where many synthetic molecules could potentially bind and modulate receptor activity.
Activation mechanism and approaches for modulating activity
Binding of competitive agonist to the VFT induces a series of conformational changes in all of the domains and activates the G-protein. This activation mechanism is particularly complicated as a result of the constitutive dimerization of this family. The dimeric receptor contains four or six independent domains in which allosteric interactions occur between each neighboring pair such that a conformational change in one domain will facilitate changes in others. For a long time, how the different domains work together to activate the coupled effectors remained poorly understood. The main hindrance in investigating this issue stems from difficulties in solving the receptor structure in the presence and absence of agonist. In 2000, the first crystal structures of the mGlu1 receptor broke this barrier. These structures revealed the dynamics of the VFT and mechanism of modulation by glutamate. A subsequent study in 2002 reported the structure of the mGlu1 receptor in the presence of an antagonist (MCPG) or an allosteric modulator (Gd3+). However, the structure of the 7TM domain remains unsolved, and details of the conformational change of the complete receptor in response to stimulation remain elusive. Our current knowledge regarding the activation process relies mainly on bioinformatic analyses, mutation constructs and advanced functional techniques. In general, the activation progress of class C GPCRs includes the following three sequential events: 1) a competitive agonist binds to one VFT in the dimer and stabilizes the closed conformation; 2) the VFT in the closed conformation transduces the activation signal to the 7TM directly or via the CRD; 3) the rearranged 7TM activates the G protein.
Ligand recognition by VFT
Crystal structure analysis of the mGlu1 receptor revealed that agonist binding induces rearrangement of the dimeric VFTs33, 34. In the resting state (R), lobes 2 of each monomer are far away from one another and the dimeric VFTs are in an open conformation. In the active state (A), lobe 2 from each monomer moves close enough to contact each other, while the dimeric VFTs are stabilized in a closed conformation in response to binding of a competitive agonist. Consistent with the above model, N-glycan wedge scanning in the GABAB receptor revealed that the interaction and relative movement of lobe 2 from each monomer is important for agonist affinity and receptor activation42.
Constitutive dimerization ensures that each receptor dimer contains two orthosteric sites in most cases. However, according to mutational analysis in mGlu receptors, one ligand is sufficient to activate the receptor dimer48. That is, a ligand binding to one subunit leads to the closure of one VFT and Aco (Active/closed open) conformation is sufficient to stabilize the active conformation of the receptor. Although the Acc (Active/close close) conformation with two bound agonists has higher activation efficacy, a cation such as Gd3+ is needed to stabilize this conformation. In the absence of a cation, electrostatic repulsion between lobe 2 from each monomer would make this conformation drastically unstable34.
Activating signal transduction from the VFT to the 7TM
Because the VFT and the 7TM of class C GPCRs are relatively independent domains, transduction of the activating signal from the VFT to the 7TM is a crucial step in receptor activation, despite the fact that many details remain to be defined. For mGlu-like receptors, the CRD plays a central role in transmitting the activating signal from the VFT to the 7TM. A conserved disulfide bond between the VFT and the CRD is indispensable for the allosteric interaction between the two domains46. Furthermore, a recent observation showed that ligand binding to the VFT triggers the relative movement of two CRDs during receptor activation. The introduction of an inter-subunit disulfide bond between the two CRDs in the receptor dimer stabilized the active conformation49. For the GABAB receptor, which lacks the CRD, it is most likely that the VFT directly interacts with the 7TM, independent of the short linker between them19.
The model that one VFT is capable of activating one receptor dimer raises the question of whether the closed VFT domain activates the 7TM domain in the same subunit (cis-activation) or the closed VFT activates the 7TM of the other subunit (trans-activation). Recently, it has been demonstrated that the 7TM of the obligatory heterodimeric GABAB receptor can be directly trans-activated either by the GABAB1 VFT and 7TM or by the dimeric VFTs formed by GABAB1 and GABAB250. In contrast, only cis-activation occurs in the T1R receptor31. The activation mechanism of homodimeric receptors with two orthosteric sites is difficult to study. The results for mGlu-like receptors showed that both cis- and trans-activation occur in the mGlu receptor activation mechanism51.
G-protein activation by 7TM
Due to the difficulties in transmembrane protein research, no crystal structures of the 7TM domain of any class C GPCR have been determined. There is no direct data to indicate that a conformational change in the 7TM occurs during receptor activation. Bioinformatic and mutational analyses suggest that the 7TM oscillates between various active and inactive conformations46, 47. FRET detection of the conformational change in the iloops of the mGlu1 receptor demonstrated that agonist binding induces iloop2 from each monomer in the dimeric 7TMs to move apart from each other, implying that the dimeric 7TMs rotate away from the interface during the activation process52, 53, 54.
Chimeric constructs and mutational analyses indicate that the C-terminal end of the Gα subunit lies within a cavity formed by iloop 2 and 3 in class C GPCRs55. In addition, the 8th helix also plays a role in G-protein coupling55, 56. Finally, it is important to mention that the highly conserved and un-usually short iloop 3 of class C GPCRs plays an equivalent role to that of iloop 2 in rhodopsin-like receptors57.
Ligand recognition sites
Class C GPCRs contain multiple ligand interaction sites as a result of their particularly complicated structure and activation mechanism. These ligand binding sites are divided into two groups: orthosteric and allosteric binding sites. The endogenous ligand binding sites, or orthosteric sites, reside in the VFT domain. Both competitive agonists and antagonists interact with this site and induce significant conformational changes in the VFT: binding of a full agonist stabilizes a closed conformation35, whereas binding of competitive antagonists stabilizes an open conformation33, 34, 43. Binding of partial agonists results in a partial or a complete, yet unstable, closure of the VFT domain35, 58. In contrast, the allosteric sites are topographically distinct from the orthosteric sites in any given receptor. The binding of allosteric modulators changes the receptor conformation and, thereby, the affinities and/or efficacies of orthosteric ligands. In general, the positive allosteric modulators (PAMs) facilitate the action of the orthosteric agonists, whereas the negative allosteric modulators (NAMs) block the activation of orthosteric agonists by stabilizing the 7TM in an inactive conformation. The large extracellular domain and constitutive dimerization of class C GPCRs provide more potential allosteric sites compared with other GPCRs. To date, there are three groups of allosteric sites in class C GPCRs that have been reported (Figure 3).

Great variety of ligands to modulate class C GPCRs function. (A) Schematic model of orthosteric sites and allosteric sites in mGlu-like receptor. There are four groups of allosteric sites in class C GPCR: sites in the 7TM, which have been studied extensively; sites in the extracellular domain including VFT and CRD and sites in the interface between VFT, CRD and 7TM, the latter two open the new possibilities to modulate activity of mGlu receptors. The structure model was built according to the crystal structure of mGlu3 VFT and CRD (PDB ID 2E4W) and the crystal structure of bovine rhodopsin 7TM (PDB ID 1GZM). (B) Modulation mechanism of various ligands on the function of homodimeric class C GPCRs. The orthosteric agonists promote the VFT closure while the antagonists prevent it. The extracellular domain allosteric modulators (EDAM) bind to a site adjacent to the orthosteric site and increase the agonist effect. The Gd3+ ion binds to the interface of the lobe 2 of the VFTs and stabilizes the full active conformation when both VFTs are closed. The sweet proteins brazzein or monellin interact on the CRD of human T1R3 and increase the agonist effect. The typical PAMs or NAMs bind to the 7TM and stabilize the active or inactive conformation of 7TM, respectively.
7TM allosteric sites
Due to the existence of the large extracellular domain, the 7TM of class C GPCRs lacks an orthosteric site, which instead is located within the VFT. However, the binding pocket is conserved and is formed by residues in TM3, 5, 6, and 7, which correspond to orthosteric sites within the 7TM of rhodopsin-like receptors3, 59. Many allosteric modulators for class C GPCRs have been demonstrated to bind in this pocket. Homology modeling, docking analysis and mutagenesis studies have shown that nine conserved amino acid residues in the 7TM of T1R3 are involved in allosteric modulator binding. The corresponding residues have also been found in the 7TM of CaS60, 61, 62, 63 and mGlu receptors64, 65, 66. This implies that class C GPCRs share a common binding site for allosteric modulators. Distinct from this common binding pocket, there are several other allosteric sites located in the 7TM of class C GPCRs. Taken together, the main group of allosteric sites in class C GPCRs resides in the 7TM. Most allosteric modulators that have been described for class C GPCRs interact with this domain.
VFT allosteric sites adjacent to orthosteric sites
Recently, the VFT binding pocket was shown to be large enough to accommodate both orthosteric and allosteric sites, which are adjacent to each other but do not overlap. Small molecules binding to this allosteric site could cooperate with endogenous ligand to stabilize the closed conformation of the VFT. These small molecules are new allosteric modulators and are called extracellular domain allosteric modulators (EDAM). To date, there are three groups of EDAMs and their corresponding sites have been identified: IMP to the T1R1 VFT of the umami taste receptor31, SE-2/SE-3 to the T1R2 VFT of the sweet taste receptor67 and the (R)-PCEP derivatives with long alkyl chains to the VFT of the mGlu4 receptor68, 69.
Allosteric sites located at the interfaces between the VFT, CRD and 7TM
Constitutive dimerization plays a crucial role in the activation of class C GPCRs so the sites involved in dimerization represent another group of allosteric sites. In the Acc conformation of the mGlu1 receptor, electrostatic repulsion from the four adjacent negatively charged residues Glu233 and Glu238 (and the analogous residues in the dimeric VFTs) makes the active conformation unstable. The introduction of a cation, such as Gd3+ or Ca2+, can neutralize this electrostatic repulsion and stabilize the active conformation. It was shown that the Gd3+ ion binds at the interface between lobe 2 of the VFTs34. Therefore, the interface of the dimeric VFT constitutes a group of allosteric sites.
Recent data show that the relative movement of dimeric CRDs is potentially involved in the mGlu receptor activation process49, so this region could represent another allosteric site. In support of this hypothesis, Jiang et al identified 10 residues in the CRD of human T1R3 and the hinge region of T1R2 that play an important role in the effect of sweet proteins, such as brazzein45.
Ligand binding sites of four typical class C GPCR family members
So far, there are only two therapeutic drugs that target class C GPCRs on the market: Baclofen, an agonist targeting the GABAB receptor, and Cinacalcet, an allosteric modulator targeting the CaS receptor. The former represents a conventional orthosteric drug, while the latter represents a novel allosteric modulator. Allosteric modulators currently attract significant attention because they offer important advantages over orthosteric drugs. First, they often have no effect on their own and act in concert with physiological receptor activation, which results in fewer side effects and a decreased propensity for desensitization; second, their binding sites do not undergo selective pressure so they display a higher subtype selectivity; third, multiple allosteric sites make it easy to synthesize novel molecules that exhibit increased bioavailability and desirable pharmacokinetic properties. Diverse allosteric modulators have been identified for class C GPCRs as a result of their plentiful allosteric sites and the numerous possibilities to modulate their function by acting on multiple steps during the activation process (Table 1).
Taste receptors — multiplicity of various ligand-binding sites
A unique characteristic of taste receptors is their diversity of ligand-binding sites. Aside from the orthosteric sites, there are at least eight allosteric sites that have been identified in taste receptors: the EDAM sites for IMP in T1R1-VFT31 and for SE-2/SE-3 in T1R2-VFT67; the allosteric agonist sites for sweet proteins in T1R3-CRD45, cyclamate in sweet receptor T1R3 7TM70, S807 in T1R1 7TM31 and S819 in T1R2-7TM31; the PAM site for cyclamate in the umami receptor T1R3-7TM70; and the NAM site for lactisole in both the sweet and umami receptor T1R3-7TM71. These sites might represent potential targets for health-related products or drugs to treat diseases, such as hypertension or diabetes.
mGlu receptors — the most promising candidates for clinical applications
The orthosteric sites of mGlu receptor subtypes are the most highly conserved throughout evolution, such that there are almost no orthosteric ligands that display higher selectivity for a given subtype. Moreover, the glutamate-binding pocket strictly selects for agonists with amino acid-like structures, which are notoriously difficult to synthesize and display undesirable pharmacokinetics. By contrast, most of the allosteric modulators for mGlu receptors possess better subtype selectivity as a result of less conserved allosteric sites and better pharmacological properties due to their structural diversity and more extensive lipophilic nature72.
The first allosteric modulator that was discovered for class C GPCRs is CPCCOEt, which functions as a NAM for the mGlu1 receptor. Numerous allosteric modulators of group I mGlu receptors have since been identified. It has been proposed that the movement of Trp798 in TM6 of mGlu1 (Trp784 at the homologous position in mGlu5) is essential for receptor activation66. The PAMs stabilize the active conformation of this group by facilitating the movement of a conserved Trp in TM6, whereas the NAMs prevent the relative movement between TM6 and TM366. For the mGlu5 receptor, most PAMs and NAMs share an overlapping binding pocket that is composed of TM3, 5, 6, and 773, except for a small number of distinct sites74. For the mGlu1 receptor, however, the PAMs and NAMs bind to distinct sites in the 7TM64, 65, 75, 76, 77, except for a shared site that consists of Val757 in TM365, 77. Removed from the conserved binding pocket, there is a distinct allosteric site located in TM1. An unique PAM for both the mGlu1 and mGlu5 receptors, CPPHA, was shown to bind to this site. Phe585 in TM1 of mGlu5 (Phe599 at the corresponding position in mGlu1) is essential for the recognition of CPPHA74.
The allosteric modulators of the mGlu5 receptor are leading with regard to the development of pharmaceuticals that target class C GPCRs. Convincing preclinical data have shown a significant effect of several PAMs in schizophrenia14. Furthermore, positive clinical results have also been obtained for NAMs in L-DOPA-induced tardive dyskinesia in Parkinson's disease11.
Most allosteric modulators for group II mGlu receptors are PAMs. These modulators provide greater subtype selectivity compared with the agonists, especially in the case of the mGlu2 receptor. Ser688 and/or Gly689 in TM4 and Asn735 in TM5 have been shown to be involved in PAM binding to the mGlu2 receptor78. The competitive agonists for group II mGlu receptors display potent activity against anxiety79 disorders and schizophrenia80 in clinical trials; however, they are unable to discriminate between the group II subtypes. PAMs with selectivity for the mGlu2 receptor have displayed similar effects as agonists in an animal model81, which suggests that there is a high possibility for success in clinical trials.
Compared with the modulators that have been described for group I and II mGlu receptors, notably fewer allosteric modulators have been identified that target group III mGlu receptors. It also important to note that some allosteric modulators that target group I mGlu receptors have the opposite effect on group III mGlu receptors. Recently, the mGlu4 receptor has been the focus of significant attention because the corresponding PAMs that target this receptor represent promising novel drugs with which to treat Parkinson's disease11.
GABAB receptors — the unique PAM CGP7930
Currently there is only one drug on the market, baclofen, that functions as a competitive agonist towards the GABAB receptor. Clinical applications over the course of several decades have shown that baclofen is an undesirable antispastic agent due to its potent side effects, unfavorable pharmacokinetic properties and a tendency for patients to develop tolerance to the drug. The newly described allosteric modulators provide opportunities to develop new therapeutic agents for several GABAB receptor related disorders.
CGP7930 is a typical PAM that was the first to be identified that targets the GABAB receptor82. This PAM can both enhance the potency and the maximal response that is induced by GABA82. Radioligand binding experiments suggest that CGP7930 not only promotes agonist affinity to the orthosteric sites but also strengthens the interaction between the GABAB receptor and the preferred Gαo83. A growing body of evidence has shown that CGP7930 interacts with the GABAB2 7TM82. According to a recently proposed model in which agonist binding induces the widening of the cleft between the two 7TMs without changing the helical configuration of each subunit, it is possible that CGP7930 binds at the interface to enhance the separation of the two 7TMs84. Interestingly, CGP7930 has been found to function as an independent partial agonist in cAMP assays85. We reported for the first time that CGP7930 itself could induce ERK1/2 phosphorylation in cultured cerebellar granule neurons (CGNs)86. Furthermore, we found that CGP7930 alone could protect CGNs from apoptosis via transactivation of the insulin-like growth factor 1 (IGF-1) receptor22. There is no obvious difference between CGP7930 and GABA or baclofen to explain the function described above22. So far, CGP7930 is a unique PAM in that it is the only such modulator that has been reported to exert an independent physiological effect. In addition to CGP7930, a number of PAMs that target the GABAB receptor have been reported, such as GS3978387. Several allosteric agonists have also been synthesized, including rac-BHFF and its analogs88. However, no NAMs that target the GABAB receptor have been described.
It has been reported recently that several amino acids89, 90 and Ca2+91, 92 can modulate GABAB receptor function via a conserved pocket that is located near the orthosteric sites, which is reminiscent of the modulation that has been observed for mGlu-like receptors. Unfortunately, animal models suggest that the existing allosteric modulators for the GABAB receptor are not suitable for clinical use due to their low potency and unfavorable pharmacokinetic properties5. Therefore, it is necessary to identify new allosteric sites on the GABAB receptor that may lead to the discovery of new types of therapeutic ligands.
CaS receptors — first clinical success
To date, four groups of ligands have been identified for the CaS receptor: the endogenous cations93, the L-amino acids (such as L-phenylalanine and L-tryptophan94), the calcimimetics and the calcilytics95. Except for the cations, the latter three groups are all allosteric modulators. Both the orthosteric site for Ca2+ and the allosteric site for L-amino acids reside in the VFT. The amino acid sites are adjacent to the orthosteric site, which corresponds to the amino acid binding pocket in the mGlu or GABAB receptors96. The calcimimetic and calcilytic sites are located in the 7TM. Structurally similar calcimimetics and calcilytics share a common allosteric binding pocket, whereas structurally distinct calcilytics recognize distinct sites97.
Among these ligands, the orthosteric agonists are inorganic ions, so it is difficult to mimic them with synthetic molecules. The L-amino acid type modulators are also not suitable for therapeutic development due to their poor bioavailability and blood-brain barrier permeability. The calcimimetics and the calcilytics, however, have successfully circumvented these problems. The calcimimetic drug Cinacalcet has already been approved to treat hyperparathyroidism clinically; meanwhile, several calcilytics have shown potent effects in animal models of osteoporosis27 and hypocalcemia26. Although one calcilytic drug, ronacaleret, did not display positive effects in a phase II clinical trial98, a second generation of calcilytics with optimized characteristics is awaiting clinical validation.
Conclusion
Class C GPCRs distinguish themselves from other GPCRs by two distinct structural features: first, they possess an unusually large extracellular domain that is responsible for orthosteric ligand recognition, while the 7TM (which normally contains the orthosteric ligand-binding site) has gained many allosteric sites; second, the functional class C GPCR molecules are obligatory dimers, so the interface between the VFT, CRD, and 7TM constitutes another important allosteric site. Furthermore, it was recently demonstrated that the VFT is large enough to accommodate allosteric modulatory sites adjacent to the orthosteric sites. The unique structure and complicated activation mechanism of class C GPCRs makes it possible to modulate their function by many new approaches. In recent years, allosteric modulation has become the most attractive approach because of the decreased side effects and development of patient tolerance, improved subtype selectivity and increased chemical accessibility. The development of allosteric modulators for class C GPCRs has progressed fast. Among them, Cinacalcet was the first clinical success. Following Cinacalcet, group I and II mGlu receptor modulators are expected to enter the market in the near future as the next generation of drugs that target class C GPCRs. By contrast, allosteric drugs that modulate the group III mGlu and GABAB receptors might represent a drug generation for the more distant future. To promote the application of allosteric modulation therapeutics that target class C GPCRs, future efforts should focus on investigating the precise structural dynamics and allosteric modulation mechanisms. Determination of the receptor structures is a direct way to address such issues. Traditional mutational analysis and chimeric constructs are also powerful tools that report on related information in the absence of a crystal structure for a particular receptor. Advanced functional assays, such as BRET (bioluminescence resonance energy transfer) or FRET (fluorescence resonance energy transfer), are widely used to reveal conformational changes and dimer or oligomer formation. Additionally, computational approaches, such as ligand- or structure-based homology modeling and docking, are gaining importance as valuable complements to experimental structure-function studies. These techniques, in combination with modern drug screening assays, make it possible to identify molecules targeting class C GPCRs through sites and mechanisms other then traditional orthosteric small molecules.
References
George SR, O'Dowd BF, Lee SP . G-protein-coupled receptor oligomerization and its potential for drug discovery. Nat Rev Drug Discov 2002; 1: 808–20.
Lagerstrom MC, Schioth HB . Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat Rev Drug Discov 2008; 7: 339–57.
Pin JP, Galvez T, Prezeau L . Evolution, structure, and activation mechanism of family 3/C G-protein-coupled receptors. Pharmacol Ther 2003; 98: 325–54.
Rondard P, Goudet C, Kniazeff J, Pin JP, Prezeau L . The complexity of their activation mechanism opens new possiblities for the modulation of mGlu and GABABclass C G protein-coupled receptors. Neuropharmacology 2011; 60: 82–92.
Urwyler S . Allosteric modulation of family C G-protein-coupled receptors: from molecular insights to therapeutic perspectives. Pharmacol Rev 2011; 63: 59–126.
Kniazeff J, Prezeau L, Rondard P, Pin JP, Goudet C . Dimers and beyond: The functional puzzles of class C GPCRs. Pharmacol Ther 2011; 130: 9–25.
Conn PJ, Christopoulos A, Lindsley CW . Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat Rev Drug Discov 2009; 8: 41–54.
Conn PJ . Pin J-P . Pharmacology and functions of metabotropic glutamate receptors. Annue Rev Pharmacol Toxicol 1997; 37: 205–37.
Niswender CM, Conn PJ . Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annue Rev Pharmacol Toxicol 2010; 50: 295–322.
Pin JP, Acher F . The metabotropic glutamate receptors: structure, activation mechanism and pharmacology. Curr Drug Targets CNS Neurol Disord 2002; 1: 297–317.
Johnson KA, Conn PJ, Niswender CM . Glutamate receptors as therapeutic targets for Parkinson's disease. CNS Neurol Disord Drug Targets 2009; 8: 475–91.
Dolen G, Carpenter RL, Ocain TD, Bear MF . Mechanism-based approaches to treating fragile X. Pharmacol Ther 2010; 127: 78–93.
Marino MJ . Williams DL Jr, O'Brien JA, Valenti O, McDonald TP, Clements MK, et al. Allosteric modulation of group III metabotropic glutamate receptor 4: a potential approach to parkinson's disease treatment. Proc Natl Acad Sci U S A 2003; 100: 13668–73.
Conn PJ, Lindsley CW, Jones CK . Activation of metabotropic gluatamate receptors as a novel approach for the treatment of schizophrenia. Trends Pharmacol Sci 2009; 30: 25–31.
Bettler B, Tiao JY . Molecular diversity, trafficking and subcellular localization of GABAB receptors. Pharmacol Ther 2006; 110: 533–43.
Jones KA, Borowsky B, Tamm JA, Craig DA, Durkin MM, Dai M, et al. GABAB receptors function as heteromeric assembly of the subunits GABABR1 and GABABR2. Nature 1998; 396: 674–79.
Kaupmann K, Malitschek B, Schuler V, Heid J, Froestl W, Beck P, et al. GABAB-receptor subtypes assemble into functional heteromeric complexes. Nature 1998; 396: 683–7.
Galvez T, Parmentier ML, Joly C, Malitschek B, Kaupmann K, Kuhn R, et al. Mutagenesis and modeling of the GABAB receptor extracellular domain support a Venus flytrap mechanism for ligand binding. J Biol Chem 1999; 274: 13362–9.
Margeta-Mitrovic M, Jan YN, Jan LY . Function of GB1 and GB2 subunits in G protein coupling of GABAB receptors. Proc Natl Acad Sci U S A 2001; 98: 14649–54.
Kuramoto N, Wilkins ME, Fairfax BP, Revilla-Sanchez R, Terunuma M, Tamaki K, et al. Phospho-dependent functional modulation of GABAB receptors by the metabolic sensor AMP-dependent protein kinase. Neuron 2007; 53: 233–47.
Dave KR, Lange-Asschenfeldt C, Raval AP, Prado R, Busto R, Saul I, et al. Ischemic preconditioning ameliorates excitotoxicity by shifting glutamate/gamma-aminobutyric acid release and biosynthesis. J Neurosci Res 2005; 82: 665–73.
Tu H, Xu C, Zhang W, Liu Q, Rondard P, Pin JP, et al. GABAB receptor activation protects neurons from apoptosis via IGF-1 receptor transactivation. J Neurosci 2010; 30: 749–59.
Goudet C, Magnaghi V, Landry M, Nagy F . Gereau RW 4th, Pin JP . Metabotropic receptors for glutamate and GABA in pain. Brain Res Rev 2009; 60: 43–56.
Cryan JF, Kaupmann K . Don't worry 'B' happy!: a role for GABAB receptors in anxiety and depression. Trends Pharmacol Sci 2005; 26: 36–43.
Bowery NG . GABAB receptor: a site of therapeutic benefit. Curr Opin Pharmacol 2006; 6: 37–43.
Brown EM . Clinical lessons from the calcium-sensing receptor. Nat Clin Pract Endocrinol Metab 2007; 3: 122–33.
Deal C . Future therapeutic targets in osteoporosis. Curr Opin Rheumatol 2009; 21: 380–5.
Brown EM . Anti-parathyroid and anti-calcium sensing receptor antibodies in autoimmune hypoparathyroidism. Endocrinol Metab Clin North Am 2009; 38: 437–45.
Montmayeur JP, Liberles SD, Matsunami H, Buck LB . A candidate taste receptor gene near a sweet taste locus. Nat Neurosci 2001; 4: 492–8.
Nelson G, Hoon MA, Chandrashekar J, Zhang Y, Ryba NJ, Zuker CS . Mammalian sweet taste receptors. Cell 2001; 106: 381–90.
Zhang F, Klebansky B, Fine RM, Xu H, Pronin A, Liu H, et al. Molecular mechanism for the umami taste synergism. Proc Natl Acad Sci U S A 2008; 105: 20930–4.
Cao J, Huang S, Qian J, Huang J, Jin L, Su Z, et al. Evolution of the class C GPCR Venus flytrap modules involved positive selected functional divergence. BMC Evol Biol 2009; 9: 67.
Kunishima N, Shimada Y, Tsuji Y, Sato T, Yamamoto M, Kumasaka T, et al. Structural basis of glutamate recognition by a dimeric metabotropic glutamate receptor. Nature 2000; 407: 971–7.
Tsuchiya D, Kunishima N, Kamiya N, Jingami H, Morikawa K . Structural views of the ligand-binding cores of a metabotropic glutamate receptor complexed with an antagonist and both glutamate and Gd3+. Proc Natl Acad Sci U S A 2002; 99: 2660–5.
Bessis AS, Rondard P, Gaven F, Brabet I, Triballeau N, Prezeau L, et al. Closure of the Venus flytrap module of mGlu8 receptor and the activation process: Insights from mutations converting antagonists into agonists. Proc Natl Acad Sci U S A 2002; 99: 11097–102.
Kniazeff J, Saintot PP, Goudet C, Liu J, Charnet A, Guillon G, et al. Locking the dimeric GABAB G-protein-coupled receptor in its active state. J Neurosci 2004; 24: 370–7.
Tsuji Y, Shimada Y, Takeshita T, Kajimura N, Nomura S, Sekiyama N, et al. Cryptic dimer interface and domain organization of the extracellular region of metabotropic glutamate receptor subtype 1. J Biol Chem 2000; 275: 28144–51.
Romano C, Miller JK, Hyrc K, Dikranian S, Mennerick S, Takeuchi Y, et al. Covalent and noncovalent interactions mediate metabotropic glutamate receptor mGlu5 dimerization. Mol Pharmacol 2001; 59: 46–53.
Ray K, Hauschild BC, Steinbach PJ, Goldsmith PK, Hauache O, Spiegel AM . Identification of the cysteine residues in the amino-terminal extracellular domain of the human Ca2+ receptor critical for dimerization. Implications for function of monomeric Ca2+ receptor. J Biol Chem 1999; 274: 27642–50.
Ray K, Hauschild BC . Cys-140 is critical for metabotropic glutamate receptor-1 dimerization. J Biol Chem 2000; 275: 34245–51.
Liu J, Maurel D, Etzol S, Brabet I, Ansanay H, Pin JP, et al. Molecular determinants involved in the allosteric control of agonist affinity in the GABAB receptor by the GABAB2 subunit. J Biol Chem 2004; 279: 15824–30.
Rondard P, Huang S, Monnier C, Tu H, Blanchard B, Oueslati N, et al. Functioning of the dimeric GABAB receptor extracellular domain revealed by glycan wedge scanning. EMBO J 2008; 27: 1321–32.
Muto T, Tsuchiya D, Morikawa K, Jingami H . Structures of the extracellular regions of the group II/III metabotropic glutamate receptors. Proc Natl Acad Sci U S A 2007; 104: 3759–64.
Hu J, Hauache O, Spiegel AM . Human Ca2+ receptor cysteine-rich domain. Analysis of function of mutant and chimeric receptors. J Biol Chem 2000; 275: 16382–9.
Jiang P, Ji Q, Liu Z, Snyder LA, Benard LM, Margolskee RF, et al. The cysteine-rich region of T1R3 determines responses to intensely sweet proteins. J Biol Chem 2004; 279: 45068–75.
Rondard P, Liu J, Huang S, Malhaire F, Vol C, Pinault A, et al. Coupling of agonist binding to effector domain activation in metabotropic glutamate-like receptors. J Biol Chem 2006; 281: 24653–61.
Goudet C, Gaven F, Kniazeff J, Vol C, Liu J, Cohen-Gonsaud M, et al. Heptahelical domain of metabotropic glutamate receptor 5 behaves like rhodopsin-like receptors. Proc Natl Acad Sci U S A 2004; 101: 378–83.
Kniazeff J, Bessis AS, Maurel D, Ansanay H, Prezeau L, Pin JP . Closed state of both binding domains of homodimeric mGlu receptors is required for full activity. Nat Struct Mol Biol 2004; 11: 706–13.
Huang S, Cao J, Jiang M, Labesse G, Liu J, Pin JP, et al. Interdomain movements in metabotropic glutamate receptor activation. Proc Natl Acad Sci U S A 2011; 108: 15480–5.
Monnier C, Tu H, Bourrier E, Vol C, Lamarque L, Trinquet E, et al. Trans-activation between 7TM domains: implication in heterodimeric GABAB receptor activation. EMBO J 2011; 30: 32–42.
Brock C, Oueslati N, Soler S, Boudier L, Rondard P, Pin JP . Activation of a dimeric metabotropic glutamate receptor by intersubunit rearrangement. J Biol Chem 2007; 282: 33000–8.
Tateyama M, Abe H, Nakata H, Saito O, Kubo Y . Ligand-induced rearrangement of the dimeric metabotropic glutamate receptor 1alpha. Nat Struct Mol Biol 2004; 11: 637–42.
Tateyama M, Kubo Y . Dual signaling is differentially activated by different active states of the metabotropic glutamate receptor 1alpha. Proc Natl Acad Sci U S A 2006; 103: 1124–8.
Marcaggi P, Mutoh H, Dimitrov D, Beato M, Knopfel T . Optical measurement of mGluR1 conformational changes reveals fast activation, slow deactivation, and sensitization. Proc Natl Acad Sci U S A 2009; 106: 11388–93.
Pin JP, Joly C, Heinemann SF, Bockaert J . Domains involved in the specificity of G protein activation in phospholipase C-coupled metabotropic glutamate receptors. EMBO J 1994; 13: 342–8.
Bai M, Trivedi S, Brown EM . Dimerization of the extracellular calcium-sensing receptor (CaR) on the cell surface of CaR-transfected HEK293 cells. J Biol Chem 1998; 273: 23605–10.
Yamashita T, Terakita A, Shichida Y . The second cytoplasmic loop of metabotropic glutamate receptor functions at the third loop position of rhodopsin. J Biochem 2001; 130: 149–55.
Frauli M, Hubert N, Schann S, Triballeau N, Bertrand HO, Acher F, et al. Amino-pyrrolidine tricarboxylic acids give new insight into group III metabotropic glutamate receptor activation mechanism. Mol Pharmacol 2007; 71: 704–12.
Brauner-Osborne H, Wellendorph P, Jensen AA . Structure, pharmacology and therapeutic prospects of family C G-protein coupled receptors. Curr Drug Targets 2007; 8: 169–84.
Hu J, Reyes-Cruz G, Chen W, Jacobson KA, Spiegel AM . Identification of acidic residues in the extracellular loops of the seven-transmembrane domain of the human Ca2+ receptor critical for response to Ca2+ and a positive allosteric modulator. J Biol Chem 2002; 277: 46622–31.
Petrel C, Kessler A, Maslah F, Dauban P, Dodd RH, Rognan D, et al. Modeling and mutagenesis of the binding site of Calhex 231, a novel negative allosteric modulator of the extracellular Ca2+-sensing receptor. J Biol Chem 2003; 278: 49487–94.
Petrel C, Kessler A, Dauban P, Dodd RH, Rognan D, Ruat M . Positive and negative allosteric modulators of the Ca2+-sensing receptor interact within overlapping but not identical binding sites in the transmembrane domain. J Biol Chem 2004; 279: 18990–7.
Hu J, McLarnon SJ, Mora S, Jiang J, Thomas C, Jacobson KA, et al. A region in the seven-transmembrane domain of the human Ca2+ receptor critical for response to Ca2+. J Biol Chem 2005; 280: 5113–20.
Pagano A, Ruegg D, Litschig S, Stoehr N, Stierlin C, Heinrich M, et al. The non-competitive antagonists 2-methyl-6-(phenylethynyl)pyridine and 7-hydroxyiminocyclopropan[b]chromen-1a-carboxylic acid ethyl ester interact with overlapping binding pockets in the transmembrane region of group I metabotropic glutamate receptors. J Biol Chem 2000; 275: 33750–8.
Malherbe P, Kratochwil N, Knoflach F, Zenner MT, Kew JN, Kratzeisen C, et al. Mutational analysis and molecular modeling of the allosteric binding site of a novel, selective, noncompetitive antagonist of the metabotropic glutamate 1 receptor. J Biol Chem 2003; 278: 8340–7.
Malherbe P, Kratochwil N, Zenner MT, Piussi J, Diener C, Kratzeisen C, et al. Mutational analysis and molecular modeling of the binding pocket of the metabotropic glutamate 5 receptor negative modulator 2-methyl-6-(phenylethynyl)-pyridine. Mol Pharmacol 2003; 64: 823–32.
Zhang F, Klebansky B, Fine RM, Liu H, Xu H, Servant G, et al. Molecular mechanism of the sweet taste enhancers. Proc Natl Acad Sci U S A 2010; 107: 4752–7.
Triballeau N, Acher F, Brabet I, Pin JP, Bertrand HO . Virtual screening workflow development guided by the “receiver operating characteristic” curve approach. Application to high-throughput docking on metabotropic glutamate receptor subtype 4. J Med Chem 2005; 48: 2534–47.
Selvam C, Oueslati N, Lemasson IA, Brabet I, Rigault D, Courtiol T, et al. A virtual screening hit reveals new possibilities for developing group III metabotropic glutamate receptor agonists. J Med Chem 2010; 53: 2797–813.
Jiang P, Cui M, Zhao B, Snyder LA, Benard LM, Osman R, et al. Identification of the cyclamate interaction site within the transmembrane domain of the human sweet taste receptor subunit T1R3. J Biol Chem 2005; 280: 34296–305.
Jiang P, Cui M, Zhao B, Liu Z, Snyder LA, Benard LM, et al. Lactisole interacts with the transmembrane domains of human T1R3 to inhibit sweet taste. J Biol Chem 2005; 280: 15238–46.
Gregory KJ, Dong EN, Meiler J, Conn PJ . Allosteric modulation of metabotropic glutamate receptors: structural insights and therapeutic potential. Neuropharmacology 2011; 60: 66–81.
Muhlemann A, Ward NA, Kratochwil N, Diener C, Fischer C, Stucki A, et al. Determination of key amino acids implicated in the actions of allosteric modulation by 3,3'-difluorobenzaldazine on rat mGlu5 receptors. Eur J Pharmacol 2006; 529: 95–104.
Chen Y, Goudet C, Pin JP, Conn PJ . N-{4-Chloro-2-[(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)methyl]phenyl}-2-hy droxybenzamide (CPPHA) acts through a novel site as a positive allosteric modulator of group 1 metabotropic glutamate receptors. Mol Pharmacol 2008; 73: 909–18.
Litschig S, Gasparini F, Rueegg D, Stoehr N, Flor PJ, Vranesic I, et al. CPCCOEt, a noncompetitive metabotropic glutamate receptor 1 antagonist, inhibits receptor signaling without affecting glutamate binding. Mol Pharmacol 1999; 55: 453–61.
Knoflach F, Mutel V, Jolidon S, Kew JN, Malherbe P, Vieira E, et al. Positive allosteric modulators of metabotropic glutamate 1 receptor: characterization, mechanism of action, and binding site. Proc Natl Acad Sci U S A 2001; 98: 13402–7.
Hemstapat K, de Paulis T, Chen Y, Brady AE, Grover VK, Alagille D, et al. A novel class of positive allosteric modulators of metabotropic glutamate receptor subtype 1 interact with a site distinct from that of negative allosteric modulators. Mol Pharmacol 2006; 70: 616–26.
Rowe BA, Schaffhauser H, Morales S, Lubbers LS, Bonnefous C, Kamenecka TM, et al. Transposition of three amino acids transforms the human metabotropic glutamate receptor (mGluR)-3-positive allosteric modulation site to mGluR2, and additional characterization of the mGluR2-positive allosteric modulation site. J Pharmacol Exp Ther 2008; 326: 240–51.
Grillon C, Cordova J, Levine LR . Morgan CA 3rd . Anxiolytic effects of a novel group II metabotropic glutamate receptor agonist (LY354740) in the fear-potentiated startle paradigm in humans. Psychopharmacology (Berl) 2003; 168: 446–54.
Patil ST, Zhang L, Martenyi F, Lowe SL, Jackson KA, Andreev BV, et al. Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: a randomized Phase 2 clinical trial. Nat Med 2007; 13: 1102–7.
Swanson CJ, Bures M, Johnson MP, Linden AM, Monn JA, Schoepp DD . Metabotropic glutamate receptors as novel targets for anxiety and stress disorders. Nat Rev Drug Discov 2005; 4: 131–44.
Urwyler S, Mosbacher J, Lingenhoehl K, Heid J, Hofstetter K, Froestl W, et al. Positive allosteric modulation of native and recombinant gamma-aminobutyric acid(B) receptors by 2,6-Di-tert-butyl-4-(3-hydroxy-2,2-dimethyl-propyl)-phenol (CGP7930) and its aldehyde analog CGP13501. Mol Pharmacol 2001; 60: 963–71.
DeLapp NW . The antibody-capture [35S]GTPgammaS scintillation proximity assay: a powerful emerging technique for analysis of GPCR pharmacology. Trends Pharmacol Sci 2004; 25: 400–1.
Matsushita S, Nakata H, Kubo Y, Tateyama M . Ligand-induced rearrangements of the GABAB receptor revealed by fluorescence resonance energy transfer. J Biol Chem 2010; 285: 10291–9.
Binet V, Brajon C, Le Corre L, Acher F, Pin JP, Prezeau L . The heptahelical domain of GABAB2 is activated directly by CGP7930, a positive allosteric modulator of the GABAB receptor. J Biol Chem 2004; 279: 29085–91.
Tu H, Rondard P, Xu C, Bertaso F, Cao F, Zhang X, et al. Dominant role of GABAB2 and Gbetagamma for GABAB receptor-mediated-ERK1/2/CREB pathway in cerebellar neurons. Cell Signal 2007; 19: 1996–2002.
Urwyler S, Pozza MF, Lingenhoehl K, Mosbacher J, Lampert C, Froestl W, et al. N,N′-Dicyclopentyl-2-methylsulfanyl-5-nitro-pyrimidine-4,6-diamine (GS39783) and structurally related compounds: novel allosteric enhancers of gamma-aminobutyric acidB receptor function. J Pharmacol Exp Ther 2003; 307: 322–30.
Malherbe P, Masciadri R, Norcross RD, Knoflach F, Kratzeisen C, Zenner MT, et al. Characterization of (R,S)-5,7-di-tert-butyl-3-hydroxy-3-trifluoromethyl-3H-benzofuran-2-one as a positive allosteric modulator of GABAB receptors. Br J Pharmacol 2008; 154: 797–811.
Kerr DI, Ong J, Puspawati NM, Prager RH . Arylalkylamines are a novel class of positive allosteric modulators at GABAB receptors in rat neocortex. Eur J Pharmacol 2002; 451: 69–77.
Kerr DI, Ong J . Potentiation of metabotropic GABAB receptors by L-amino acids and dipeptides in rat neocortex. Eur J Pharmacol 2003; 468: 103–8.
Wise A, Green A, Main MJ, Wilson R, Fraser N, Marshall FH . Calcium sensing properties of the GABAB receptor. Neuropharmacology 1999; 38: 1647–56.
Galvez T, Urwyler S, Prezeau L, Mosbacher J, Joly C, Malitschek B, et al. Ca2+ requirement for high-affinity gamma-aminobutyric acid (GABA) binding at GABAB receptors: involvement of serine 269 of the GABABR1 subunit. Mol Pharmacol 2000; 57: 419–26.
Brauner-Osborne H, Jensen AA, Sheppard PO, O'Hara P, Krogsgaard-Larsen P . The agonist-binding domain of the calcium-sensing receptor is located at the amino-terminal domain. J Biol Chem 1999; 274: 18382–6.
Zhang Z, Qiu W, Quinn SJ, Conigrave AD, Brown EM, Bai M . Three adjacent serines in the extracellular domains of the CaR are required for L-amino acid-mediated potentiation of receptor function. J Biol Chem 2002; 277: 33727–35.
Saidak Z, Brazier M, Kamel S, Mentaverri R . Agonists and allosteric modulators of the calcium-sensing receptor and their therapeutic applications. Mol Pharmacol 2009; 76: 1131–44.
Silve C, Petrel C, Leroy C, Bruel H, Mallet E, Rognan D, et al. Delineating a Ca2+ binding pocket within the Venus flytrap module of the human calcium-sensing receptor. J Biol Chem 2005; 280: 37917–23.
Arey BJ, Seethala R, Ma Z, Fura A, Morin J, Swartz J, et al. A novel calcium-sensing receptor antagonist transiently stimulates parathyroid hormone secretion in vivo. Endocrinology 2005; 146: 2015–22.
Fitzpatrick LA, Smith PL, McBride TA, Fries MA, Hossain M, Dabrowski CE, et al. Ronacaleret, a calcium-sensing receptor antagonist, has no significant effect on radial fracture healing time: Results of a randomized, double-blinded, placebo-controlled Phase II clinical trial. Bone 2011; 49: 845–52.
Acknowledgements
This work was supported by grants from the Key Project of National Natural Science Foundation of China (NSFC) (No 31130028) and the National Basic Research Program of China (973 Program) (No 2012CB518006).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Chun, L., Zhang, Wh. & Liu, Jf. Structure and ligand recognition of class C GPCRs. Acta Pharmacol Sin 33, 312–323 (2012). https://doi.org/10.1038/aps.2011.186
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2011.186
Keywords
This article is cited by
-
Hyaluronic acid–GPRC5C signalling promotes dormancy in haematopoietic stem cells
Nature Cell Biology (2022)
-
Bivalent ligands promote endosomal trafficking of the dopamine D3 receptor-neurotensin receptor 1 heterodimer
Communications Biology (2021)
-
GABAB receptor pas de deux: insights from high-resolution structures
Cell Research (2020)
-
Structural basis for adhesion G protein-coupled receptor Gpr126 function
Nature Communications (2020)
-
FET-based nanobiosensors for the detection of smell and taste
Science China Life Sciences (2020)
