
Abstract
Adenosine Monophosphate-activated Protein Kinase (AMPK), a serine/threonine kinase and a member of the Snf1/AMPK protein kinase family, consists of three protein subunits that together make a functional enzyme. AMPK, which is expressed in a number of tissues, including the liver, brain, and skeletal muscle, is allosterically activated by a rise in the AMP: ATP ratio (ie in a low ATP or energy depleted state). The net effect of AMPK activation is to halt energy consuming (anabolic) pathways but to promote energy conserving (catabolic) cellular pathways. AMPK has therefore often been dubbed the “metabolic master switch”. AMPK also plays a critical physiological role in the cardiovascular system. Increasing evidence suggest that AMPK might also function as a sensor by responding to oxidative stress. Mostly importantly, AMPK modulates endogenous antioxidant gene expression and/or suppress the production of oxidants. AMPK promotes cardiovascular homeostasis by ensuring an optimum redox balance on the heart and vascular tissues. Dysfunctional AMPK is thought to underlie several cardiovascular pathologies. Here we review this kinase from its structure and discovery to current knowledge of its adaptive and maladaptive role in the cardiovascular system.
Similar content being viewed by others

Introduction
One of the many important cellular control systems is the AMP-activated protein kinase (AMPK), which is the putative metabolic or energy sensor of the cell. AMPK signaling appears to have broad implications in cardiovascular health and disease. This review will focus on AMPK's emerging role as an integrating metabolic sensor in the cardiovascular system.
The discovery of AMPK is not recent. Its existence was uncovered by two independent observations reported in 1973 with the discovery that the same kinase inactivates 3-hydroxy-3-methylglutaryl coenzyme A (CoA) reductase and acetyl-CoA carboxylase (ACC) in hepatic fat metabolism1, 2. Several years after these seminal reports, Munday et al, in the process of studying the Vmax of ACC, proposed the name AMP-activated protein kinase as the primary enzyme responsible for attenuating this parameter3. Finally, with Carling et al reporting that the HMG-CoA reductase (HMGR) and ACC kinases were one and the same enzyme, the name was formally adopted in 19894, 5. Subsequently, AMPK was purified and its subunit structure was analyzed by Grahame Hardie's group at the University of Dundee6 with detailed analysis of its all important catalytic subunit published by Bruce Kemp's laboratory at the St Vincent's Medical Research Institute in Victoria, Australia7. Based on these ground breaking studies and work by various other investigators, it has been revealed that AMPK is a heterotrimer with α, β, and γ subunits. The α subunit has catalytic activity while the other two have a regulatory role. Overall, multiple AMPK subunit isoform combinations have been identified and these subunits are encoded by distinct genes. Thus far, two α subunits, two β subunits and three γ subunits have been identified8, 9, 10.
AMPK physiology
AMPK subunits
The standardized nomenclature of AMPK subunit genes utilizes a prefix PRKA followed by the subunit identifier A1, A2, B1, B2, G1, G2, and G3 (eg, PRKAG3)11. The gene loci for the subunits are located on 5 different chromosomes: α1 (5p12), α2 (1q31), β1 (12q24.1), β2 (1q21.1), γ1 (12q12–14), γ2 (7q35–36), and γ3 (2q35). With this plethora of subunits it is not surprising that gene expression and variant splicing can give rise to twelve possible heterotrimeric combinations of AMPK12 (Figure 1).

Features of the AMPK subunits (modified from Steinberg and Kemp, Physiol Rev 89: 1025–78). Colored regions are ones whose structure is known. Numbers associated with α and β subunits are N- and C- terminal residues from the crystal structure. AIS: Auto inhibitory sequence. β-SID: β-subunit interacting domain. GBD: Glycogen binding domain. αγ-SBS: α and γ subunit interacting sequence. In the expanded glycogen binding domain schematic of the β subunits, letters with numbers are sites of sugar binding (pS108 is the site at Serine 108 where phosphorylation occurs). In the γ subunit, D90, R171, D245 and D317 are residues that form H-bonds with 2′,3′-ribose hydroxyl groups, while R70, H151, R152, K170, H298 and R299 represent basic residues that occupy the solvent accessible core of the subunit which makes contact with the nucleotide phosphates.
Both α subunits are similar in that they have about 550 residues (Figure 1) as well as conserved NH2-terminal catalytic domains. The β subunits differ in the first 65 residues but in all other respects are highly conserved. The γ subunits on the other hand (and in contrast to the other two), differ in length (γ1 being the shortest at 331, γ3 intermediate at 489 and γ2 the longest at 569)11. However, all three share a COOH-terminal having about 300 residues. Significant differences exist in AMPK subunit structure and genetic sequence between mammals and yeast, for example (multiple α and γ subunits in mammals and 2 rather than three β subunits in yeast).
Evidence suggests that variance of the α subunits determines subcellular localization of the molecule with the α1 isoform being largely cytosolic as well as being associated with the plasma membrane in carotid body type 1 cells and airway epithelial cells13, 14. In contrast α2 appears to be concentrated in the nuclei of several cell types such as pancreatic β cells, neurons and skeletal muscle15, 16, 17.
The β subunits feature the glycogen binding domain (GBD) which occupies a position on the central conserved region of the subunit. The crystal structure of the GBD was reported in 200518. Another conserved region on this subunit is in the C-terminal region and there is compelling evidence that the C-terminal domain is all that is needed to form a functional αβγ unit that can be regulated by AMP19.
The three γ subunits have variable N-terminal regions followed by four tandem repeats of a 60-aa sequence named as a CBS (cystathionine β-synthase) motif by Bateman et al20. It has since been discovered that these are actually two domains on the subunit (Figure 1; Bateman 1 and 2 domains), each with the capacity to bind AMP with a 1:1 stoichiometry21. The critical nature of these domains was revealed when investigators reported attenuated AMP binding and activation when mutations were induced in these regions21. The Bateman domains also bind ATP antagonistically, but with a lower affinity than that for AMP21 and this is consistent with the fact that ATP antagonizes activation of AMPK by AMP22. Interestingly, the two Bateman domains also act cooperatively in that the second site remains inaccessible to AMP until the nucleotide has bound to the first21. This synergy between the two domains is a potential mechanism by which AMPK activation can respond to even small changes in cellular AMP levels12.
In summary, the α subunit provides catalytic activity while the β and γ subunits are regulatory. The α subunit has a Thr172 phosphorylation site. The β subunit has scaffold-like properties and also possesses myristoylation, phosphorylation and glycogen-binding sites. The γ subunit features the nucleotide-binding module. Thus, for correct function all three units act in concert and are necessary for normal function.
AMPK activation
Mammalian AMPK is sensitive to the AMP: ATP ratio. It is therefore activated as a consequence of any cellular process, normal or anomalous, that either decreases ATP levels, or increases AMP concentrations. For example, mechanisms such as hypoxia, glucose deprivation or metabolic inhibition of ATP synthesis, will all activate AMPK23. If ATP production remains unaltered but consumption is increased, the same result will ensue. Examples of increased ATP synthesis include activation of motor proteins, activity of ion channels/pumps and utilization by biosynthetic pathways. In addition, less well understood yet empirically established modulators of AMPK activity have appeared in literature and the list is ever growing. Such modulators include cytokines [leptin, adiponectin, ghrelin, cannabinoids, IL-624, ciliary neutrotrophic factor (CNTF; 25)], certain drugs (metformin26, thiazolidinediones27) and some plant derived compounds (berberine28, resveratrol29), to name a few.
Activation of AMPK complexes that contain the α1 subunit isoform are reported to be localized in the cytosol. In contrast, AMPKα2 activation results in translocation to the nucleus and is thought to facilitate modulation of gene expression30, 31. The β subunit also appears to be involved in determining the localization fate of the molecule in that myristoylation of the β subunit targets the complex to the Golgi while phosphorylation on various residues promotes nuclear translocation32.
Once AMPK is activated, it switches on (the concept of the “metabolic master switch”; most likely first proposed by Prof HARDIE of the University of Dundee) catabolic pathways that can generate ATP while at the same time, terminates processes that consume ATP. The rapid “switching” required to closely and quickly regulate and balance cellular energy resources is achieved by brisk phosphorylation of metabolic enzymes as well as that of various transcription factors and co-activators which control gene expression23. Analysis has shown that the nucleotide AMP, allosterically binds to and activates the γ subunit of AMPK which triggers phosphorylation of the α subunit at Thr172.
Upstream AMPK kinases
By the early 2000s, it had become clear that a critical phosphorylation event took place on the α subunit (on Thr172) in the process of AMPK activation. However, the identities of upstream phosphotransferases that were responsible for this had remained elusive. In 2003, breakthrough discoveries in the yeast system (Schizosaccharomyces pombe) identified Sak1 (Snf1-activating kinase-1), Elm-1 (elongated morphology-1) and Tos3 (Target of Sbf3) as kinases upstream of the Snf1 complex (homolog of the serine/threonine protein kinase found in S cerevisiae)33, 34, 35. Although unequivocal human orthologs of these three kinases have not been found in the human genome, the two protein kinases closest in sequence to these are LKB1 (a serine/threonine tumor suppressor kinase) and the Ca2+/calmodulin-dependent protein kinase kinase β (CaMKKβ)30. Evidence now demonstrates phosphorylation of the AMPK α subunit can either be dependent on, or independent of, its LKB1 activity. Specifically, LKB1 appears to be critically involved in the activation of AMPKα2 but not AMPKα130, 36. AICAR (5-Aminoimidazole-4-carboxyamide ribonucleoside; an important activator of AMPK) is an adenosine analog taken up by muscle and phosphorylated to form 5-aminoimidazole-4-carboxamide-1-D-ribofuranosyl-5'-monophosphate (ZMP), which stimulates AMPK activity and glucose transport in skeletal muscle. LKB1 is essential for AICAR induced activation of AMPK. Empirically, it has been reported that deletion of LKB1 will prevent activation of AMPKα2 in cardiac and skeletal muscle cells36.
Unlike LKB1, CaMKKβ is regulated within the cell and its levels increase in response to elevations in intracellular Ca2+ ([Ca2+]i). Therefore, stimuli that amplify ([Ca2+]i (such as bradykinin and thrombin), also activate AMPKα1 consequent upon increased CaMKKβ activity37, 38.
Downstream targets of AMPK
Once activated, AMPK can influence several downstream targets in the cell. Many of these targets are currently recognized20, however more are being discovered and it has been speculated that this number may eventually rise into the hundreds. These downstream effectors of AMPK influence diverse cellular processes and include lipid metabolism [eg, acetyl-CoA carboxylase (ACC); HMG-CoA reductase], carbohydrate metabolism (eg, glycogen synthase; 6-phosphofructo-2-kinase); cell signaling [eg, endothelial NO synthase (eNOS); insulin receptor substrate-1 (IRS-1)], ion transport [cystic fibrosis transmembrane conductance regulator (CFTR)] and transcription [eg, p300; hepatocyte nuclear factor-4α (HNF4-α); transducer of regulated CREB activity 2 (TORC2)]14, 39, 40, 41, 42, 43, 44, 45. One of the most intriguing questions has been: How does AMPK recognize its downstream targets? AMPK has been found to phosphorylate a serine residue in these targets. Further, phosphorylation sites appear to have conserved motifs where hydrophobic residues are found 5 residues from the N terminal and 4 from the C terminal (P-5 and P+4). This motif is designated Φ-[β.X]-X-X-S/T-X-X-X-Φ, where Φ is hydrophobic and β is basic. This motif has been confirmed using variant synthetic peptide substrates46, 47.
AMPK in cardiovascular physiology
Homeostatic mechanisms in the heart and the vascular endothelium are critical in maintaining cardiovascular health. These mechanisms are involved in diverse cardiovascular functions like regulation of vascular tone, maintenance of tissue perfusion, vascular permeability, myocardial function, anticoagulant activity and inflammatory responses48, 49, 50.
AMPK in the heart
While AMPK signaling has a specific physiological role in the heart, its importance is accentuated under conditions that place a stress on this organ. Such stressors include excess hemodynamic load, myocardial ischemia and hypoxia51. Specifically, AMPK activates the glycolytic pathway by phosphorylating phosphofructokinase-2, enhances fatty acid β-oxidation and enhances ATP availability52, 53. It also promotes the translocation of glucose transporter 4 (GLUT-4) to the plasma membrane in the cardiomyocyte thus increasing the uptake of glucose to serve as the primary energy substrate51.
AMPK in endothelial cells
Endothelial cells in the CVS can sense changes in hemodynamic forces, ambient pO2 and local blood-borne signals. They can then respond with appropriate control and regulatory processes to maintain homeostasis. Such responses can include release of paracrine mediators such as nitric oxide (NO), prostacyclin and endothelin-1 (ET-1)48. In addition, activity of cell surface enzymes can be modulated such as angiotensin converting enzyme (ACE) which regulates the bioactivity of vasculoactive molecules like angiotensin II and bradykinin48. Furthermore cell surface adhesion molecules are also modulated in addition to promotion of immune cell recruitment and migration in case of vascular injury along with increased vascular permeability and intravascular thrombosis48.
The central signaling molecule involved in endothelial function is nitric oxide (NO) which is synthesized from L-arginine by NO synthase (NOS) in endothelial cells. NOS exists as three isoforms: neuronal (nNOS), inducible (iNOS) and endothelial (eNOS). All three isoforms share a carboxyl terminal domain homologous to cytochrome P-450 reductase and has binding sites for nicotinamide adenine dinucleotide phosphate-oxidase (NADPH), Flavin mononucleotide (FMN) and tetrahydrobiopterin (BH4)54. eNOS is typically activated by an increase in [Ca2+]i. A strong physiological stimulus for NO synthesis vie eNOS activation is the shear stress caused by increases in blood flow54. It has also been demonstrated that other physiological stimuli such as insulin, estrogen and vascular endothelial growth factor (VEGF) can activate the phosphatidyl inositol-3 kinase (PI3K)/Akt system and in turn this phosphorylates NOS et Ser117755. AMPK is the only kinase recognized to date that can also potentially phosphorylate eNOS on more than one site. These sites have been reported as Ser1177 and Ser63341, 56 (activating sites on the reductase domain of eNOS), and at Thr495 (inhibitory at the CaM-binding domain of the enzyme)41. Many reports have identified that AMPK-dependent eNOS activation (at Ser1177) can occur following endothelial cell stimulation by diverse agents and these include VEGF57, peroxisome proliferator-activated receptors γ (PPAR-γ) agonists58, AICAR59, and metformin60. In 2004, it was reported that the biguanide drug, metformin activated AMPK mediated by mitochondrial RNS (reactive nitrogen species) and the PI3K pathway61. Specifically, this report demonstrated that metformin activated AMPK and increased the phosphorylation of ACC (its downstream effector) at Ser79 in cultured BAEC (bovine aortic endothelial cells), that this was mediated through c-Src and was PI3K-dependent, was ONOO−-dependent, that the peroxynitrile oxidant was sourced to the mitochondrion and that inhibition of mitochondrial complex I activated AMPK. Further, the paper also validated these findings in vivo. Further studies have also established that metformin-induced AMPK activation is beneficial to endothelial function via Heat Shock Protein 90 (hsp90) mediated activation of eNOS60. These investigations have revealed that metformin can increase the conversion of arginine into citrulline in BAECs in a dose-dependent fashion, suggesting that NO synthesis was occurring via activation of eNOS. This study also revealed that eNOS activation by metformin was PI3K-dependent, was clearly mediated through the activation of AMPK and that it enhanced the association of hsp90 (an important stress response marker) with eNOS. Recently, evidence has also emerged that NO itself might act as an endogenous activator of AMPK62. In this report, we have shown that NO activates AMPK in endothelial cells through a Ca2+-dependent mechanism involving CaMKKβ and that AMPK activation can itself increase NO release through AMPK-dependent phosphorylation of eNOS at Ser1177. These data imply that a positive feedback relationship might exist between eNOS and AMPK activation. However, further investigation of this possibility is warranted.
Once NO is synthesized via the activation of eNOS it diffuses to the surrounding tissue and exerts its multiple physiological effects which include vascular smooth muscle relaxation and proliferation, and inhibiting leukocyte adhesion and migration, platelet adhesion and aggregation and expression of adhesion molecules63, 64. In disease conditions, the endothelium undergoes complex changes and loses its protective and pro-homeostatic function and acquires pro-atherosclerotic properties. Collectively these abnormalities are referred to as “endothelial dysfunction” and are typified by reduced bioavailability of NO65. In these alterations, AMPK also appears to play an important role.
AMPK and vascular smooth muscle
AMPK activation in the endothelium is also linked to endothelial control of vascular smooth muscle function, particularly vasorelaxation by NO which appears to be regulated, at least in part by AMPK activity. Vasorelaxation is a critical arm of vascular tone and tone is a central determinant of blood pressure regulation. Sustained high blood pressure is responsible for a variety of serious disease states such as hypertrophic cardiomyopathy, coronary artery insufficiency, myocardial infarction, hypertensive encephalopathy, cerebrovascular disease, hypertensive retinopathy and hypertensive nephropathy being important examples. It has been established that metformin therapy is beneficial for the cardiovascular system by virtue of its ability to improve vasodilatory function66, 67. It is possible that these effects of metformin are dependent on eNOS activation as demonstrated by various investigators. Another possibility that merits consideration is the fact that endothelium-dependent vasorelaxation is not exclusively regulated by NO. It is provocative to speculate that AMPK might be linked to vasorelaxation via the generation of epoxyeicosatrienoic acids by the cytochrome P450 epoxygenases30. In fact LKB1 and AMPK can be activated by stimulation of the constitutive androstane receptor and pregnane X receptor (both of the nuclear receptor superfamily that are thought to be involved in the detoxification of xenobiotics), using Phenobarbital (a classic P450 inducer)68. In mice that lack hepatic α1 and α2 isoforms of AMPK, this response is not detectable69. In keeping with the vasorelaxative role of AMPK, it is now fairly well established that AICAR and metformin can both relax arterial preparations ex vivo60, 61, 70. Interestingly, there seems to be a species- and vessel-dependent difference in published literature in terms of chemical sensitivity of AMPK activation. For example, unlike rat and mouse aortae, porcine carotid artery smooth muscle is reportedly insensitive to AICAR and metformin, even though this tissue can have AMPK activation by other means, such as by hypoxia and 2-deoxyglucose (which causes a metabolic block in the glycolytic pathway)71.
AMPK as a redox sensor and modulator
Oxidative stress and a shift in the cellular redox balance are critical underpinnings of endothelial dysfunction. In turn, endothelial disturbances underlie cardiovascular pathology30. The decreased bioavailability of NO is also associated with the generation of reactive oxygen species (ROS) in the vessel wall such as peroxynitrite72. Many studies have now established that there is an intricate balance between AMPK signaling and the redox balance in the vascular milieu. For example, AMPK has been shown to inhibit the formation of reactive oxygen species (ROS) by NADPH oxidase and stimulate NO production be eNOS30. Furthermore, AMPK has also been implicated in JNK activation, NF-κB-mediated transcription, E-selectin expression and vascular cell adhesion molecule-1 (VCAM-1) expression, in endothelial cells that have been exposed to H2O2, TNF-α or fatty acids. As a consequence, these signaling events lead to attenuated monocyte adhesion to the endothelial surface73, 74, 75, 76. Silencing AMPKα1 has also been reported to decrease the expression of Manganese Superoxide Dismutase (MnSOD), catalase, γ-glutamylcysteine synthase and thioredoxin, in endothelial cells77. AMPK also appears to have direct links to NADPH oxidase, a membrane bound enzyme complex which is normally latent in neutrophils and is activated to assemble in the membranes during the respiratory burst. It generates O2·− transferring electrons from NADPH inside the cell across the membrane and coupling these to molecular oxygen to produce O2·−. It has been shown that AICAR or AMP can suppress the production of superoxide anion stimulated by phorbol esters or fMLP (Formyl-Methionyl-Leucyl-Phenylalanine)78. In neutrophils, AICAR also has the ability to reduce PMA-dependent (phorbol 12-myristate 13-acetate; a potent activator of PKC) H2O2 release and induction of phosphorylation of JNK, p38, MAPK, and ERK1/230. The exposure of cultured human endothelial cells (HEC) to high glucose concentration (10 mmol/L) can generate ROS and this can effectively be attenuated by AMPK-activating drugs, such as rosiglitazone79. AMPK can also influence the cellular redox state via prevention of tyrosine nitration and inhibition of prostacyclin synthase in endothelial cells that have been exposed to high glucose. This has been reported with the demonstration that AICAR could inhibit high glucose-induced nitration and inactivation of prostacyclin synthase in HUVECs, that AMPK activation was necessary for AICAR to reduce oxidative stress, that prostacyclin synthase nitration by AMPK was mediated through an upregulation of UCP-2 (mitochondrial uncoupling protein-2) and that this was dependent on activation of p38 kinase as well80. These findings are especially of note given that the basic role of uncoupling proteins is to prevent oxidative injury or minimize oxidative stress. In fact, such a mechanism has also been validated in in vivo experiments where AICAR-mediated AMPK activation markedly increases UCP-2 expression and reduction of O2·− and prostacyclin synthase nitration in diabetic wild-type but not in AMPKα-deleted mice80. Unpublished recent data (Xu et al) from our laboratory has also underscored the role of UCP-2 in mitigating mitochondrial ROS synthesis in an AMPK-dependent manner under ischemic conditions in skeletal vasculature. In this study we have shown that abrogation of ROS production via UCP-2 is critical for the compensatory neovascularization that is an important element in tissue response to ischemia/hypoxia.
Recently, novel findings demonstrating that hypoxia-reperfusion via ONOO− can activate AMPK in a c-Src mediated, PI3K-dependent manner, have been reported81. These data also show that the thromboxane receptor (TPr), once stimulated, can trigger ROS-mediated LKB1-dependent AMPK activation in vascular smooth muscle resulting in inhibition of cellular protein synthesis82, that PKCζ can regulate AMPK activity by increasing phosphorylation at Ser428 of LKB1 (resulting in association of LKB1 with AMPK and consequent phosphorylation of LKB1 itself at Thr172)83, that the PKCζ phosphorylation of LKB1 results in nuclear export of the kinase and prompts AMPK activation in endothelial cells84, and most recently, ROS are required in the process of AMPK activation by statin drugs as well as PKCζ85. A new mechanism has also been proposed through which H2O2 can mediate AMPK activation independent of changes in the AMP:ATP ratio in endothelial cells. This has been observed in this cell type under nutritional and oxidative stress with free radical species appearing to be derived from the mitochondria. The most interesting aspect of this recent study is that under nutritive challenge (utilizing 2-deoxyglucose to induce a biochemical block by preventing glycolytic breakdown at the hexokinase step), AMPK activation led to autophagy rather than cell death. Further, under conditions of hypoxia, endothelial cell death was prevented in an AMPK and ROS dependent manner. Overall, several recent studies reinforce the idea that AMPK activation is acutely sensitive to cellular redox balance and this relationship appears to be independent of its putative function of responding to ATP depletion (or more precisely elevated AMP:ATP) and triggering energy conserving pathways in the cell. AMPK's response to the redox balance also appears to favor cell survival pathways.
AMPK and cardiovascular diseases
Myocardial hypertrophy
Cardiac hypertrophy can develop under physiological conditions as is seen in trained athletes as a compensatory response to intense exercise. It can also occur pathologically in response to left ventricular pressure or volume overload as seen in hypertension or valvular heart disease86. In recent years, evidence has begun to emerge that AMPK can modulate the development of cardiac hypertrophy. For example, Tian et al have reported that following aortic banding in rats that led to cardiac hypertrophy, concomitantly also activated AMPK in the myocardium87. Allard et al have also reported that hypertrophied rodent hearts exhibit increased glycolysis in conjunction with AMPK activation88. However, contrary data has also been published that suggests that cardiac hypertrophy may or may not be associated with a change in AMPK signaling. For example, phenylepherine treated hypertrophic cardiomyocytes have been shown to have unchanged AMPK activity89. More specifically, data from Dyck's group at the University of Alberta has shown depressed AMPK activity in a spontaneously hypertensive rat model86. Furthermore, mice lacking adiponectin that have reduced AMPK signaling are more susceptible to afterload-induced hypertrophy90. Interestingly, reports suggest that pharmacological activation of AMPK can mitigate the hypertrophy associated with hemodynamic pressure overload. For example, treatment with the AMPK activator AICAR has been shown to reverse cardiac hypertrophy in rats following aortic banding91. Resveratrol (a phytoalexin that can potently activate AMPK) is also reported to inhibit hypertrophy in the heart following aortic banding in the rat92. However, it has to be pointed out that AMPK does not appear to have a physiological role in limiting the hypertrophic response of cardiac myocytes based on the findings of Russell et al and Xing et al. These investigators demonstrated that mouse hearts with impaired AMPK signaling are not hypertrophied93, 94.
Ischemic injury and ischemic pre-conditioning
Myocardial ischemia-reperfusion injury (inflammatory and oxidative damage to the cardiac muscle caused by reperfusion following a relief of ischemia), is an important cardiopathic mechanism where AMPK is also thought to play a role. It is known for example, that AMPKα1 and α2 activity is elevated during cardiac ischemia5. Increased AMPK activity likely compensates for the ischemic condition by upregulating GLUT4 translocation and enhancing fatty acid oxidation53, 95. However, it remains unclear if AMPK activity contributes positively or negatively to reperfusion in that reports have suggested both scenarios being dominant93, 94.
Short periods of cardiac ischemia can protect the heart against injury from subsequent and more sustained ischemia. This phenomenon is called ischemia pre-conditioning and has been known since the 1980s following a seminal description by Keith Reimer's group at Duke University96. It has been shown that AMPK can be activated during these short periods of ischemia in the heart97, 98. AICAR has been shown to potentiate pre-conditioning as well as prolonging the period over which the beneficial and protective effects of this phenomenon can last. This was demonstrated by Tsuchida et al and Burckhartt et al in rabbits undergoing coronary ligation99, 100. Furthermore, AICAR has also been shown to contribute to pre-conditioning in the rat liver against ischemic injury in vivo101. AMPK has also been noted to mediate pre-conditioning in the isolated cardiomyocyte under hypoxic conditions102.
AMPK genetic mutations in heart disease
Naturally occurring mutations in the γ2 subunit of AMPK have been identified that appear to be involved in the pathogenesis of the Wolf-Parkinson-White syndrome (WPWS), a ventricular pre-excitement anomaly that leads to cardiac arrhythmias. This condition is quite rare affecting 0.9 to 3% of the general population103. Mechanistically, the γ subunit mutation in WPWS appears to cause excessive glycogen storage and deposition in cardiomyocytes and these cells contribute to the formation of accessory excitation pathways (called the Bundle of Kent) resulting in dysrhythmias104. Several transgenic mouse models of AMPK γ mutations have been studied in recent years that demonstrate various disturbances in their cardiac phenotype. These transgenic models have not clarified the exact relationship of γ subunit mutations with glycogen storage anomalies, AMPK signaling and heart rhythm disturbances and further work is merited. For example, transgenic mice harboring two different mutations in the γ2 subunit appear to have opposing effects on AMPK activity in vivo. The N4881 mutation is reported to increase AMPK activity compared with wild type animals, whereas the R302Q mutation attenuates AMPK activity105, 106.
AMPK and vascular smooth muscle proliferation, angiogenesis and neointimal hyperplasia
In terms of the consequences of endothelial activation of AMPK, several aspects need to be borne in mind. Overall, the activation of this molecule by FGF (fibroblast growth factor), adiponectin, hypoxia and VEGF, seems to be critical for angiogenesis30, 107, 108, 109. For example, dominant negative AMPK (DN-AMPK) is able to suppress both endothelial cell migration in response to VEGF as well as in vitro differentiation into tube-like structures under hypoxic conditions107. Similarly, increased VEGF expression has been shown in muscle cells treated with AICAR leading to enhanced angiogenic repair in response to ischemic injury in the mouse hind limb. This phenomenon reportedly involves AMPK-dependent activation of p38 MAPK110. Quite recently, Leick, and colleagues have reported that an AMPK/PGC-1α-dependent mechanism is likely responsible for exercise-induced VEGF expression in skeletal muscle as well as exercise-training-induced prevention of senescent decline in VEGF protein content111. Intriguingly, AICAR has also been shown to decrease neointimal hyperplasia in the rat femoral artery denudation model linked to ERK1/2 inhibition (in part)112.
AMPK, ER stress, and atherosclerosis
The role of AMPK activation in anti-atherosclerotic processes in the vasculature is also currently under intense study and several fascinating insights have been revealed. For example, the activation of mammalian target of Rapamycin (mTOR) by oxidized low density lipoprotein (LDL) has been shown to be involved in smooth muscle cell proliferation, and AMPK activation by resveratrol can block the activation of the PI3K/Akt/mTOR/p70S6K pathways with consequent inhibition of DNA synthesis and proliferation of smooth muscle cells113. Research has recently revealed that AMPK acts as a suppressor of endoplasmic reticulum (ER) stress and that this is dependent on the α2 subunit expression of AMPK114. It has been demonstrated that reduction in AMPKα expression resulted in increased ER stress in human umbilical vein endothelial cells (HUVEC). When ER stress markers were measured in mouse aortic endothelial cells (MAEC) derived from AMPKα2 knockout mice, these were seen to be significantly higher compared with α1 knockout cells or WT controls. This implied that that the α2 isoform of the catalytic subunit of AMPK was critical for suppressing ER stress. In this study the mechanism of heightened ER stress associated with reduced activity of AMPKα2 was identified to be increased oxidation of the sarcoplasmic/endoplasmic ER Ca2+ ATPase (SERCA) pump in the ER. SERCA oxidation hampered its activity and caused increased [Ca2+]i levels. When Ca2+ chelators were used to treat endothelial cells in this study, ER stress was mitigated. Abrogation of ER stress in vivo was also achieved following long term treatment of LDL receptor/AMPKα2 double knockout mice with the potent chemical ER chaperone, Tauroursodeoxycholic acid (TUDCA)114. As part of this study it was also observed that LDLr/AMPKα2 double knockout mice that were fed a pro-atherosclerotic high fat diet, developed advanced aortic root atherosclerosis. Interestingly, histological analysis of these lesions revealed that they were significantly more severe compared with LDLr only knockout animals fed a similar diet. These findings compellingly demonstrated that AMPKα2 not only attenuated ER stress in vitro as well as in vivo, but also played a critical role in vascular pathology characterized by atherosclerosis. Thus, the role of AMPK in the development of this significant human pathology cannot be understated.
Summary and conclusions
It has become evident with continuing research that AMPK plays a pivotal role in the complex dynamics of bioenergetics, in both health and disease. The signaling system that has AMPK as its centerpiece is fairly complex. A fact that speaks to how far reaching are the effects of activation or inhibition of this kinase. In a physiologic sense, this molecule has layers of influence that it exerts on cellular processes. These include switching between anabolic and catabolic states and altering cellular dynamics by directly influencing genetic controls and protein expression. In conditions where cellular systems are under metabolic strain, AMPK's ability to quickly correct dynamic processes appears to be an evolutionarily conserved mechanism seen across a wide phylogenetic scale, indeed in all organisms studied thus far. Even when the cellular homeostatic machinery has been overrun by pathological forces, AMPK acquires a central position in buffeting these fluxes, frequently pointing them in a correcting direction and occasionally contributing to the mounting damage that leads to cell death.
Although AMPK is traditionally thought of as an intracellular energy switch or fuel gauge, recent research has shown that this molecule is also a key player in maintaining physiological processes in the cardiovascular system, both in the heart and the vasculature. In the heart, signaling by AMPK appears to regulate the bioenergetic status of the cardiomyocyte as well as maintain the heart muscle and its electrical pace makers in optimum condition. As described, mutations in the AMPK subunit structure can cause rare but serious heart rhythm anomalies. In the endothelium, AMPK appears to redress the disturbed redox balance associated with vascular pathology and also exerts anti-atherosclerotic effects via improving NO bioavailability, attenuation of ROS stress and activation of pro-angiogenic factors.
Some drugs and compounds that have become centerpieces of therapeutics as well as public health dogma, in disorders like diabetes (eg, metformin), cardiovascular disease (eg, polyphenols a resveratrol) and infectious diseases (eg, berberine), appear to deliver their protective/therapeutic effects via modulation of AMPK signaling (Figure 2).

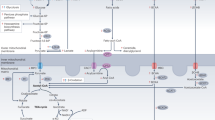
Summary of AMPK activation and its effects in the endothelium (modified from Zou and Wu, Clin Exp Pharmacol Physiol 2008 35 (5–6): 535–45). AMPK senses an increase in AMP:ATP by binding AMP to the γ subunit. This allosterically activation facilitates the activity of LKB1 of which AMPK is a substrate. Additional regulation is affected by CaMKKβ. Once activated, AMPK improves NO bioavailability, enhances FFA oxidation and inhibits ROS production in the endothelium via several different pathways. In addition, by blocking the NFκB signaling system, AMPK also inhibits inflammatory responses in the endothelium, possible contributing to its role as an anti-atherogenic regulator. Improvement in endothelial function by AMPK activation is well established in the literature. However, its direct role in preventing atherosclerosis is less well characterized.
References
Carlson CA, Kim KH . Regulation of hepatic acetyl coenzyme A carboxylase by phosphorylation and dephosphorylation. J Biol Chem 1973; 248: 378–80.
Beg ZH, Allmann DW, Gibson DM . Modulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity with cAMP and wth protein fractions of rat liver cytosol. Biochem Biophys Res Commun 1973; 54: 1362–9.
Munday MR, Campbell DG, Carling D, Hardie DG . Identification by amino acid sequencing of three major regulatory phosphorylation sites on rat acetyl-CoA carboxylase. Eur J Biochem 1988; 175: 331–8.
Carling D, Clarke PR, Zammit VA, Hardie DG . Purification and characterization of the AMP-activated protein kinase. Copurification of acetyl-CoA carboxylase kinase and 3-hydroxy-3-methylglutaryl-CoA reductase kinase activities. Eur J Biochem 1989; 186: 129–36.
Steinberg GR, Kemp BE . AMPK in Health and Disease. Physiol Rev 2009; 89: 1025–78.
Davies SP, Hawley SA, Woods A, Carling D, Haystead TA, Hardie DG . Purification of the AMP-activated protein kinase on ATP-gamma-sepharose and analysis of its subunit structure. Eur J Biochem 1994; 223: 351–7.
Mitchelhill KI, Stapleton D, Gao G, House C, Michell B, Katsis F, et al. Mammalian AMP-activated protein kinase shares structural and functional homology with the catalytic domain of yeast Snf1 protein kinase. J Biol Chem 1994; 269: 2361–4.
Stapleton D, Gao G, Michell BJ, Widmer J, Mitchelhill K, Teh T, et al. Mammalian 5′-AMP-activated protein kinase non-catalytic subunits are homologs of proteins that interact with yeast Snf1 protein kinase. J Biol Chem 1994; 269: 29343–6.
Chen Z, Heierhorst J, Mann RJ, Mitchelhill KI, Michell BJ, Witters LA, et al. Expression of the AMP-activated protein kinase beta1 and beta2 subunits in skeletal muscle. FEBS Lett 1999; 460: 343–8.
Cheung PC, Salt IP, Davies SP, Hardie DG, Carling D . Characterization of AMP-activated protein kinase gamma-subunit isoforms and their role in AMP binding. Biochem J 2000; 346: 659–69.
Steinberg GR, Kemp BE . AMPK in Health and Disease. Physiol Rev 2009; 89: 1025–78.
Towler MC, Hardie DG . AMP-activated protein kinase in metabolic control and insulin signaling. Circ Res 2007; 100: 328–41.
Evans AM, Mustard KJ, Wyatt CN, Peers C, Dipp M, Kumar P, et al. Does AMP-activated protein kinase couple inhibition of mitochondrial oxidative phosphorylation by hypoxia to calcium signaling in O2-sensing cells? J Biol Chem 2005; 280: 41504–11.
Hallows KR, Kobinger GP, Wilson JM, Witters LA, Foskett JK . Physiological modulation of CFTR activity by AMP-activated protein kinase in polarized T84 cells. Am J Physiol Cell Physiol 2003; 284: C1297–308.
Salt IP, Johnson G, Ashcroft SJ, Hardie DG . AMP-activated protein kinase is activated by low glucose in cell lines derived from pancreatic beta cells, and may regulate insulin release. Biochem J 1998; 335: 533–9.
Turnley AM, Stapleton D, Mann RJ, Witters LA, Kemp BE, Bartlett PF . Cellular distribution and developmental expression of AMP-activated protein kinase isoforms in mouse central nervous system. J Neurochem 1999; 72: 1707–16.
Ai H, Ihlemann J, Hellsten Y, Lauritzen HP, Hardie DG, Galbo H, et al. Effect of fiber type and nutritional state on AICAR- and contraction-stimulated glucose transport in rat muscle. Am J Physiol Endocrinol Metab 2002; 282: E1291–300.
Polekhina G, Feil SC, Gupta A, O'Donnell P, Stapleton D, Parker MW . Crystallization of the glycogen-binding domain of the AMP-activated protein kinase beta subunit and preliminary X-ray analysis. Acta Crystallogr Sect F Struct Biol Cryst Commun 2005; 61: 39–42.
Hudson ER, Pan DA, James J, Lucocq JM, Hawley SA, Green KA, et al. A novel domain in AMP-activated protein kinase causes glycogen storage bodies similar to those seen in hereditary cardiac arrhythmias. Curr Biol 2003; 13: 861–6.
Bateman A . The structure of a domain common to archaebacteria and the homocystinuria disease protein. Trends Biochem Sci 1997; 22: 12–3.
Scott JW, Hawley SA, Green KA, Anis M, Stewart G, Scullion GA, et al. CBS domains form energy-sensing modules whose binding of adenosine ligands is disrupted by disease mutations. J Clin Invest 2004; 113: 274–84.
Corton JM, Gillespie JG, Hawley SA, Hardie DG . 5-aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur J Biochem 1995; 229: 558–65.
Hardie DG . AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol 2007; 8: 774–85.
Ruderman NB, Keller C, Richard AM, Saha AK, Luo Z, Xiang X, et al. Interleukin-6 regulation of AMP-activated protein kinase. Potential role in the systemic response to exercise and prevention of the metabolic syndrome. Diabetes 2006; 55 (Suppl 2): S48–54.
Watt MJ, Dzamko N, Thomas WG, Rose-John S, Ernst M, Carling D, et al. CNTF reverses obesity-induced insulin resistance by activating skeletal muscle AMPK. Nat Med 2006; 12: 541–8.
Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest 2001; 108: 1167–74.
Fryer LG, Parbu-Patel A, Carling D . The Anti-diabetic drugs rosiglitazone and metformin stimulate AMP-activated protein kinase through distinct signaling pathways. J Biol Chem 2002; 277: 25226–32.
Lee YS, Kim WS, Kim KH, Yoon MJ, Cho HJ, Shen Y, et al. Berberine, a natural plant product, activates AMP-activated protein kinase with beneficial metabolic effects in diabetic and insulin-resistant states. Diabetes 2006; 55: 2256–64.
Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature 2006; 444: 337–42.
Fisslthaler B, Fleming I . Activation and signaling by the AMP-activated protein kinase in endothelial cells. Circ Res 2009; 105: 114–27.
Wojtaszewski JF, Nielsen P, Hansen BF, Richter EA, Kiens B . Isoform-specific and exercise intensity-dependent activation of 5′-AMP-activated protein kinase in human skeletal muscle. J Physiol 2000; 528 Pt 1: 221–6.
Warden SM, Richardson C . O'Donnell J Jr, Stapleton D, Kemp BE, Witters LA . Post-translational modifications of the beta-1 subunit of AMP-activated protein kinase affect enzyme activity and cellular localization. Biochem J 2001; 354: 275–83.
Hardie DG . AMPK and SNF1: Snuffing Out Stress. Cell Metab 2007; 6: 339–40.
Hong SP, Leiper FC, Woods A, Carling D, Carlson M . Activation of yeast Snf1 and mammalian AMP-activated protein kinase by upstream kinases. Proc Natl Acad Sci USA 2003; 100: 8839–43.
Sutherland CM, Hawley SA, McCartney RR, Leech A, Stark MJ, Schmidt MC, et al. Elm1p is one of three upstream kinases for the Saccharomyces cerevisiae SNF1 complex. Curr Biol 2003; 13: 1299–305.
Sakamoto K, Zarrinpashneh E, Budas GR, Pouleur AC, Dutta A, Prescott AR, et al. Deficiency of LKB1 in heart prevents ischemia-mediated activation of AMPKalpha2 but not AMPKalpha1. Am J Physiol Endocrinol Metab 2006; 290: E780–8.
Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, et al. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab 2005; 2: 9–19.
Stahmann N, Woods A, Carling D, Heller R . Thrombin activates AMP-activated protein kinase in endothelial cells via a pathway involving Ca2+/calmodulin-dependent protein kinase kinase beta. Mol Cell Biol 2006; 26: 5933–45.
Hong YH, Varanasi US, Yang W, Leff T . AMP-activated protein kinase regulates HNF4alpha transcriptional activity by inhibiting dimer formation and decreasing protein stability. J Biol Chem 2003; 278: 27495–501.
Koo SH, Flechner L, Qi L, Zhang X, Screaton RA, Jeffries S, et al. The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature 2005; 437: 1109–11.
Chen ZP, Mitchelhill KI, Michell BJ, Stapleton D, Rodriguez-Crespo I, Witters LA, et al. AMP-activated protein kinase phosphorylation of endothelial NO synthase. FEBS Lett 1999; 443: 285–9.
Clarke PR, Hardie DG . Regulation of HMG-CoA reductase: identification of the site phosphorylated by the AMP-activated protein kinase in vitro and in intact rat liver. EMBO J 1990; 9: 2439–46.
Carling D, Hardie DG . The substrate and sequence specificity of the AMP-activated protein kinase. Phosphorylation of glycogen synthase and phosphorylase kinase. Biochim Biophys Acta 1989; 1012: 81–6.
Jakobsen SN, Hardie DG, Morrice N, Tornqvist HE . 5′-AMP-activated protein kinase phosphorylates IRS-1 on Ser-789 in mouse C2C12 myotubes in response to 5-aminoimidazole-4-carboxamide riboside. J Biol Chem 2001; 276: 46912–6.
Yang W, Hong YH, Shen XQ, Frankowski C, Camp HS, Leff T . Regulation of transcription by AMP-activated protein kinase: phosphorylation of p300 blocks its interaction with nuclear receptors. J Biol Chem 2001; 276: 38341–4.
Weekes J, Ball KL, Caudwell FB, Hardie DG . Specificity determinants for the AMP-activated protein kinase and its plant homologue analysed using synthetic peptides. FEBS Lett 1993; 334: 335–9.
Dale S, Wilson WA, Edelman AM, Hardie DG . Similar substrate recognition motifs for mammalian AMP-activated protein kinase, higher plant HMG-CoA reductase kinase-A, yeast SNF1, and mammalian calmodulin-dependent protein kinase I. FEBS Lett 1995; 361: 191–5.
Li JM, Shah AM . Endothelial cell superoxide generation: regulation and relevance for cardiovascular pathophysiology. Am J Physiol Regul Integr Comp Physiol 2004; 287: R1014–30.
Shah AM . Paracrine modulation of heart cell function by endothelial cells. Cardiovasc Res 1996; 31: 847–67.
Cai H, Harrison DG . Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res 2000; 87: 840–4.
Nagata D, Hirata Y . The role of AMP-activated protein kinase in the cardiovascular system. Hypertens Res; 33: 22–8.
Marsin AS, Bertrand L, Rider MH, Deprez J, Beauloye C, Vincent MF, et al. Phosphorylation and activation of heart PFK-2 by AMPK has a role in the stimulation of glycolysis during ischaemia. Curr Biol 2000; 10: 1247–55.
Kudo N, Barr AJ, Barr RL, Desai S, Lopaschuk GD . High rates of fatty acid oxidation during reperfusion of ischemic hearts are associated with a decrease in malonyl-CoA levels due to an increase in 5′-AMP-activated protein kinase inhibition of acetyl-CoA carboxylase. J Biol Chem 1995; 270: 17513–20.
Hirata Y, Nagata D, Suzuki E, Nishimatsu H, Suzuki J, Nagai R . Diagnosis and treatment of endothelial dysfunction in cardiovascular disease. Int Heart J 2010; 51: 1–6.
Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, et al. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature 1999; 399: 597–601.
Chen Z, Peng IC, Sun W, Su MI, Hsu PH, Fu Y, et al. AMP-activated protein kinase functionally phosphorylates endothelial nitric oxide synthase Ser633. Circ Res 2009; 104: 496–505.
Reihill JA, Ewart MA, Hardie DG, Salt IP . AMP-activated protein kinase mediates VEGF-stimulated endothelial NO production. Biochem Biophys Res Commun 2007; 354: 1084–8.
Boyle JG, Logan PJ, Ewart MA, Reihill JA, Ritchie SA, Connell JM, et al. Rosiglitazone stimulates nitric oxide synthesis in human aortic endothelial cells via AMP-activated protein kinase. J Biol Chem 2008; 283: 11210–7.
Morrow VA, Foufelle F, Connell JM, Petrie JR, Gould GW, Salt IP . Direct activation of AMP-activated protein kinase stimulates nitric-oxide synthesis in human aortic endothelial cells. J Biol Chem 2003; 278: 31629–39.
Davis BJ, Xie Z, Viollet B, Zou MH . Activation of the AMP-activated kinase by antidiabetes drug metformin stimulates nitric oxide synthesis in vivo by promoting the association of heat shock protein 90 and endothelial nitric oxide synthase. Diabetes 2006; 55: 496–505.
Zou MH, Kirkpatrick SS, Davis BJ, Nelson JS, Wiles WGt, Schlattner U, et al. Activation of the AMP-activated protein kinase by the anti-diabetic drug metformin in vivo. Role of mitochondrial reactive nitrogen species. J Biol Chem 2004; 279: 43940–51.
Zhang J, Xie Z, Dong Y, Wang S, Liu C, Zou MH . Identification of nitric oxide as an endogenous activator of the AMP-activated protein kinase in vascular endothelial cells. J Biol Chem 2008; 283: 27452–61.
Luscher TF . The endothelium. Target and promoter of hypertension? Hypertension 1990; 15: 482–5.
Taddei S, Ghiadoni L, Virdis A, Versari D, Salvetti A . Mechanisms of endothelial dysfunction: clinical significance and preventive non-pharmacological therapeutic strategies. Curr Pharm Des 2003; 9: 2385–402.
Versari D, Daghini E, Virdis A, Ghiadoni L, Taddei S . Endothelial dysfunction as a target for prevention of cardiovascular disease. Diabetes Care 2009; 32 Suppl 2: S314–21.
Katakam PV, Ujhelyi MR, Hoenig M, Miller AW . Metformin improves vascular function in insulin-resistant rats. Hypertension 2000; 35: 108–12.
Sartoretto JL, Melo GA, Carvalho MH, Nigro D, Passaglia RT, Scavone C, et al. Metformin treatment restores the altered microvascular reactivity in neonatal streptozotocin-induced diabetic rats increasing NOS activity, but not NOS expression. Life Sci 2005; 77: 2676–89.
Blattler SM, Rencurel F, Kaufmann MR, Meyer UA . In the regulation of cytochrome P450 genes, phenobarbital targets LKB1 for necessary activation of AMP-activated protein kinase. Proc Natl Acad Sci USA 2007; 104: 1045–50.
Rencurel F, Foretz M, Kaufmann MR, Stroka D, Looser R, Leclerc I, et al. Stimulation of AMP-activated protein kinase is essential for the induction of drug metabolizing enzymes by phenobarbital in human and mouse liver. Mol Pharmacol 2006; 70: 1925–34.
Goirand F, Solar M, Athea Y, Viollet B, Mateo P, Fortin D, et al. Activation of AMP kinase alpha1 subunit induces aortic vasorelaxation in mice. J Physiol 2007; 581 (Pt 3): 1163–71.
Rubin LJ, Magliola L, Feng X, Jones AW, Hale CC . Metabolic activation of AMP kinase in vascular smooth muscle. J Appl Physiol 2005; 98: 296–306.
Bouloumie A, Bauersachs J, Linz W, Scholkens BA, Wiemer G, Fleming I, et al. Endothelial dysfunction coincides with an enhanced nitric oxide synthase expression and superoxide anion production. Hypertension 1997; 30: 934–41.
Schulz E, Dopheide J, Schuhmacher S, Thomas SR, Chen K, Daiber A, et al. Suppression of the JNK pathway by induction of a metabolic stress response prevents vascular injury and dysfunction. Circulation 2008; 118: 1347–57.
Hattori Y, Nakano Y, Hattori S, Tomizawa A, Inukai K, Kasai K . High molecular weight adiponectin activates AMPK and suppresses cytokine-induced NF-kappaB activation in vascular endothelial cells. FEBS Lett 2008; 582: 1719–24.
Dixit M, Bess E, Fisslthaler B, Hartel FV, Noll T, Busse R, et al. Shear stress-induced activation of the AMP-activated protein kinase regulates FoxO1a and angiopoietin-2 in endothelial cells. Cardiovasc Res 2008; 77: 160–8.
Cacicedo JM, Yagihashi N, Keaney JF Jr, Ruderman NB, Ido Y . AMPK inhibits fatty acid-induced increases in NF-kappaB transactivation in cultured human umbilical vein endothelial cells. Biochem Biophys Res Commun 2004; 324: 1204–9.
Colombo SL, Moncada S . AMPKalpha1 regulates the antioxidant status of vascular endothelial cells. Biochem J 2009; 421: 163–9.
Alba G, El Bekay R, Alvarez-Maqueda M, Chacon P, Vega A, Monteseirin J, et al. Stimulators of AMP-activated protein kinase inhibit the respiratory burst in human neutrophils. FEBS Lett 2004; 573: 219–25.
Ceolotto G, Gallo A, Papparella I, Franco L, Murphy E, Iori E, et al. Rosiglitazone reduces glucose-induced oxidative stress mediated by NAD(P)H oxidase via AMPK-dependent mechanism. Arterioscler Thromb Vasc Biol 2007; 27: 2627–33.
Xie Z, Zhang J, Wu J, Viollet B, Zou MH . Upregulation of mitochondrial uncoupling protein-2 by the AMP-activated protein kinase in endothelial cells attenuates oxidative stress in diabetes. Diabetes 2008; 57: 3222–30.
Zou MH, Hou XY, Shi CM, Kirkpatick S, Liu F, Goldman MH, et al. Activation of 5′-AMP-activated kinase is mediated through c-Src and phosphoinositide 3-kinase activity during hypoxia-reoxygenation of bovine aortic endothelial cells. Role of peroxynitrite. J Biol Chem 2003; 278: 34003–10.
Zhang M, Dong Y, Xu J, Xie Z, Wu Y, Song P, et al. Thromboxane receptor activates the AMP-activated protein kinase in vascular smooth muscle cells via hydrogen peroxide. Circ Res 2008; 102: 328–37.
Xie Z, Dong Y, Zhang M, Cui MZ, Cohen RA, Riek U, et al. Activation of protein kinase C zeta by peroxynitrite regulates LKB1-dependent AMP-activated protein kinase in cultured endothelial cells. J Biol Chem 2006; 281: 6366–75.
Xie Z, Dong Y, Scholz R, Neumann D, Zou MH . Phosphorylation of LKB1 at serine 428 by protein kinase C-zeta is required for metformin-enhanced activation of the AMP-activated protein kinase in endothelial cells. Circulation 2008; 117: 952–62.
Choi HC, Song P, Xie Z, Wu Y, Xu J, Zhang M, et al. Reactive nitrogen species is required for the activation of the AMP-activated protein kinase by statin in vivo. J Biol Chem 2008; 283: 20186–97.
Kim AS, Miller EJ, Young LH . AMP-activated protein kinase: a core signalling pathway in the heart. Acta Physiol (Oxf) 2009; 196: 37–53.
Tian R, Musi N, D'Agostino J, Hirshman MF, Goodyear LJ . Increased adenosine monophosphate-activated protein kinase activity in rat hearts with pressure-overload hypertrophy. Circulation 2001; 104: 1664–9.
Allard MF, Parsons HL, Saeedi R, Wambolt RB, Brownsey R . AMPK and metabolic adaptation by the heart to pressure overload. Am J Physiol Heart Circ Physiol 2007; 292: H140–8.
Chan AY, Soltys CL, Young ME, Proud CG, Dyck JR . Activation of AMP-activated protein kinase inhibits protein synthesis associated with hypertrophy in the cardiac myocyte. J Biol Chem 2004; 279: 32771–9.
Shibata R, Ouchi N, Ito M, Kihara S, Shiojima I, Pimentel DR, et al. Adiponectin-mediated modulation of hypertrophic signals in the heart. Nat Med 2004; 10: 1384–9.
Li HL, Yin R, Chen D, Liu D, Wang D, Yang Q, et al. Long-term activation of adenosine monophosphate-activated protein kinase attenuates pressure-overload-induced cardiac hypertrophy. J Cell Biochem 2007; 100: 1086–99.
Juric D, Wojciechowski P, Das DK, Netticadan T . Prevention of concentric hypertrophy and diastolic impairment in aortic-banded rats treated with resveratrol. Am J Physiol Heart Circ Physiol 2007; 292: H2138–43.
Russell RR, 3rd, Li J, Coven DL, Pypaert M, Zechner C, Palmeri M, et al. AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. J Clin Invest 2004; 114: 495–503.
Xing Y, Musi N, Fujii N, Zou L, Luptak I, Hirshman MF, et al. Glucose metabolism and energy homeostasis in mouse hearts overexpressing dominant negative alpha2 subunit of AMP-activated protein kinase. J Biol Chem 2003; 278: 28372–7.
Russell RR, 3rd, Bergeron R, Shulman GI, Young LH . Translocation of myocardial GLUT-4 and increased glucose uptake through activation of AMPK by AICAR. Am J Physiol 1999; 277: H643–9.
Reimer KA, Murry CE, Yamasawa I, Hill ML, Jennings RB . Four brief periods of myocardial ischemia cause no cumulative ATP loss or necrosis. Am J Physiol 1986; 251: H1306–15.
Baron SJ, Li J, Russell RR, 3rd, Neumann D, Miller EJ, Tuerk R, et al. Dual mechanisms regulating AMPK kinase action in the ischemic heart. Circ Res 2005; 96: 337–45.
Nishino Y, Miura T, Miki T, Sakamoto J, Nakamura Y, Ikeda Y, et al. Ischemic preconditioning activates AMPK in a PKC-dependent manner and induces GLUT4 up-regulation in the late phase of cardioprotection. Cardiovasc Res 2004; 61: 610–9.
Burckhartt B, Yang XM, Tsuchida A, Mullane KM, Downey JM, Cohen MV . Acadesine extends the window of protection afforded by ischaemic preconditioning in conscious rabbits. Cardiovasc Res 1995; 29: 653–7.
Tsuchida A, Yang XM, Burckhartt B, Mullane KM, Cohen MV, Downey JM . Acadesine extends the window of protection afforded by ischaemic preconditioning. Cardiovasc Res 1994; 28: 379–83.
Peralta C, Bartrons R, Serafin A, Blazquez C, Guzman M, Prats N, et al. Adenosine monophosphate-activated protein kinase mediates the protective effects of ischemic preconditioning on hepatic ischemia-reperfusion injury in the rat. Hepatology 2001; 34: 1164–73.
Sukhodub A, Jovanovic S, Du Q, Budas G, Clelland AK, Shen M, et al. AMP-activated protein kinase mediates preconditioning in cardiomyocytes by regulating activity and trafficking of sarcolemmal ATP-sensitive K(+) channels. J Cell Physiol 2007; 210: 224–36.
Rosner MH, Brady WJ Jr, Kefer MP, Martin ML . Electrocardiography in the patient with the Wolff-Parkinson-White syndrome: diagnostic and initial therapeutic issues. Am J Emerg Med 1999; 17: 705–14.
Gollob MH . Glycogen storage disease as a unifying mechanism of disease in the PRKAG2 cardiac syndrome. Biochem Soc Trans 2003; 31: 228–31.
Arad M, Moskowitz IP, Patel VV, Ahmad F, Perez-Atayde AR, Sawyer DB, et al. Transgenic mice overexpressing mutant PRKAG2 define the cause of Wolff-Parkinson-White syndrome in glycogen storage cardiomyopathy. Circulation 2003; 107: 2850–6.
Sidhu JS, Rajawat YS, Rami TG, Gollob MH, Wang Z, Yuan R, et al. Transgenic mouse model of ventricular preexcitation and atrioventricular reentrant tachycardia induced by an AMP-activated protein kinase loss-of-function mutation responsible for Wolff-Parkinson-White syndrome. Circulation 2005; 111: 21–9.
Nagata D, Mogi M, Walsh K . AMP-activated protein kinase (AMPK) signaling in endothelial cells is essential for angiogenesis in response to hypoxic stress. J Biol Chem 2003; 278: 31000–6.
Ouchi N, Kihara S, Arita Y, Nishida M, Matsuyama A, Okamoto Y, et al. Adipocyte-derived plasma protein, adiponectin, suppresses lipid accumulation and class A scavenger receptor expression in human monocyte-derived macrophages. Circulation 2001; 103: 1057–63.
Webler AC, Michaelis UR, Popp R, Barbosa-Sicard E, Murugan A, Falck JR, et al. Epoxyeicosatrienoic acids are part of the VEGF-activated signaling cascade leading to angiogenesis. Am J Physiol Cell Physiol 2008; 295: C1292–301.
Ouchi N, Shibata R, Walsh K . AMP-activated protein kinase signaling stimulates VEGF expression and angiogenesis in skeletal muscle. Circ Res 2005; 96: 838–46.
Leick L, Hellsten Y, Fentz J, Lyngby SS, Wojtaszewski JF, Hidalgo J, et al. PGC-1alpha mediates exercise-induced skeletal muscle VEGF expression in mice. Am J Physiol Endocrinol Metab 2009; 297: E92–103.
Nagata D, Takeda R, Sata M, Satonaka H, Suzuki E, Nagano T, et al. AMP-activated protein kinase inhibits angiotensin II-stimulated vascular smooth muscle cell proliferation. Circulation 2004; 110: 444–51.
Brito PM, Devillard R, Negre-Salvayre A, Almeida LM, Dinis TC, Salvayre R, et al. Resveratrol inhibits the mTOR mitogenic signaling evoked by oxidized LDL in smooth muscle cells. Atherosclerosis 2009; 205: 126–34.
Dong Y, Zhang M, Liang B, Xie Z, Zhao Z, Asfa S, et al. Reduction of AMP-activated protein kinase alpha2 increases endoplasmic reticulum stress and atherosclerosis in vivo. Circulation 2010; 121: 792–803.
Acknowledgements
We thank all members of Dr ZOU's laboratory for their contributions to the work included in this review. The work of Dr Ming-Hui ZOU's laboratory is supported by NIH grants (HL079584, HL074399, HL080499, HL089920, and HL096032), and by research awards from the American Diabetes Association, Juvenile Diabetes Research Foundation, Oklahoma Center for Advancement of Science and Technology, and a Travis Endowed Chair in Endocrinology, University of Oklahoma Health Sciences Center. Dr Ming-Hui ZOU is a recipient of a National Established Investigator Award from the American Heart Association.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Shirwany, N., Zou, MH. AMPK in cardiovascular health and disease. Acta Pharmacol Sin 31, 1075–1084 (2010). https://doi.org/10.1038/aps.2010.139
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2010.139
Keywords
This article is cited by
-
Mitochondria homeostasis: Biology and involvement in hepatic steatosis to NASH
Acta Pharmacologica Sinica (2022)
-
AMPKα2 controls the anti-atherosclerotic effects of fish oils by modulating the SUMOylation of GPR120
Nature Communications (2022)
-
Smooth muscle-specific HuR knockout induces defective autophagy and atherosclerosis
Cell Death & Disease (2021)
-
A spotlight on underlying the mechanism of AMPK in diabetes complications
Inflammation Research (2021)
-
LncRNA SNHG1 regulates vascular endothelial cell proliferation and angiogenesis via miR-196a
Journal of Molecular Histology (2020)
