
Abstract
Aim:
To design novel bifunctional derivatives of huperzine B (HupB) based on the concept of dual binding site of acetylcholinesterase (AChE) and evaluate their pharmacological activities for seeking new drug candidates against Alzheimer's disease (AD).
Methods:
Novel 16-substituted bifunctional derivatives of HupB were synthesized through chemical reactions. The inhibitory activities of the derivatives toward AChE and butyrylcholinesterase (BuChE) were determined in vitro by modified Ellman's method. Cell viability was quantified by the reduction of MTT.
Results:
A new preparative method was developed for the generation of 16-substituted derivatives of HupB, and pharmacological trials indicated that the derivatives were multifunctional cholinesterase inhibitors targeting both AChE and BuChE. Among the derivatives tested, 9c, 9e, 9f, and 9i were 480 to 1360 times more potent as AChE inhibitors and 370 to 1560 times more potent as BuChE inhibitors than the parent HupB. Further preliminary pharmacological trials of derivatives 9c and 9i were performed, including examining the mechanism of AChE inhibition, the substrate kinetics of the enzyme inhibition, and protection against hydrogen peroxide (H2O2)-induced cytotoxicity in PC12 cells.
Conclusion:
Preliminary pharmacological evaluation indicated that 16-substituted derivatives of HupB, particularly 9c and 9i, would be potentially valuable new drug candidates for AD therapy, and further exploration is needed to evaluate their pharmacological and clinical efficacies.
Similar content being viewed by others
Introduction
Alzheimer's disease (AD) is a progressive neurodegenerative disorder characterized by the presence of numerous senile plaques and neurofibrillary tangles in the brain1. It has become increasingly clear that the major pathological processes contributing to AD involve multiple causes, including genetic, environmental and endogenous factors. Therefore, AD has been viewed as one of the most enigmatic and problematic issues in biomedicine. However, recent studies have indicated that multifunctional compounds might provide greater efficacy as potential drugs for the treatment of neurodegenerative disease compared to those of single target compounds2. In fact, numerous studies have shown that acetylcholinesterase (AChE) inhibitors, the current mainstay of symptomatic therapy for AD that includes donepezil, galantamine, rivastigmine and huperzine A (HupA), are able to attenuate neuronal damage and death induced by cytotoxic insults, and might consequently slow down or even prevent AD pathogenesis3. It is therefore probable that future AD treatments will involve multifunctional approaches4. In addition to synthetic ligands, naturally occurring multipotent agents for AD have also attracted scientists' attention5.
Crystallographic and biochemical studies of AChE have revealed two potential binding sites in the active-site gorge. These include the catalytic anionic site and the peripheral anionic site, which are located at the bottom and top of the deep gorge of the enzyme, respectively. The active site is critically involved in hydrolysis of neurotransmitter acetylcholine (ACh), and the peripheral site plays a role in mediating β-amyloid (Aβ) peptide aggregation6, 7, which is a hallmark of the AD patient's brain. According to previous studies, AChE inhibitors targeting both binding sites would be expected to exert additional benefits besides ameliorating cognitive deficits by enhancing synaptic ACh levels8, 9.
Natural (–)-huperzine B (HupB, 1, Figure 1), a Lycopodium alkaloid isolated from the Chinese medicinal herb Huperzia serrata10, has been demonstrated as an effective and reversible inhibitor of AChE. Although the inhibitory activity of HupB is less potent than that of HupA (2), which has now been approved as a new drug for the symptomatic treatment of AD in China11, 12, HupB exhibits a higher therapeutic index than that of HupA and other positive benefits from pharmacological trials13, 14, 15, 16, 17. Therefore, HupB appears to be a promising lead compound for developing novel multifunctional AChE inhibitors for AD therapy.

Structural formulas of HupB and HupA.
Should HupB be a lead compound for AD therapy, it will be necessary to enhance the potency for AChE inhibition by preparing derivatives. We studied dual binding site derivatives starting from natural HupB several years ago. A series of HupB derivatives has been synthesized so as to bind to the dual sites of the enzyme, and remarkably, most of them demonstrate stronger AChE inhibition with an enhancement in potency of 2–3 orders of magnitude compared with the parent HupB18, 19, 20. To date, all bis- and bifunctional derivatives of HupB have been derived from the secondary amino group in HupB moiety. However, the X-ray crystallographic structure of Torpedo californica AChE complexed with HupB has demonstrated that the secondary amine participates in the interaction between the enzyme and the ligand21.
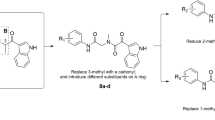
Herein, we present the rational design and preparation of novel HupB derivatives without masking the secondary amine so as to understand the action results of the amino group in the inhibitory activities of these derivatives. In this paper, we describe the synthesis, inhibitory activities and preliminary pharmacological results regarding a series of 16-substituted derivatives of HupB, all of which are shown in Scheme 1. Their main structural characteristic is that HupB moiety from the 16-position is connected through a tether chain with a terminal aromatic ring, whose π-system should allow for binding to the peripheral site of AChE8, 9. Therefore, these novel bifunctional substituted derivatives of HupB provide a new kind of heterodimeric ligand to bind and inhibit AChE.

Synthesis of 16-substituted bifunctional derivatives of HupB. Reagents and conditions: (a) trifluoroacetic anhydride, pyridine, CH2Cl2, 0 °C, 10 h. (b) methyl iodide, silver carbonate, CHCl3, RT, 24 h. (c) selenium dioxide, 1,4-dioxane, refluxing, 8 h. (d) sec-monoamine, sodium borohydride, titanium (IV) isopropoxide, EtOH, RT. (e) trimethylsilyl chloride, sodium iodide, CH3CN, refluxing, 10 h, then refluxing in MeOH, 10 h.
Materials and methods
Chemistry
Natural HupB was obtained from Wenling Pharmaceutical Factory, Zhejiang, China. Chemical reagents of analytical or chemical pure were purchased from commercial available companies and used without purification. Anhydrous solvents were obtained by distillation from sodium. Column chromatography separation was performed on silica gel (200−300 mesh).
All 16-substituted derivatives of HupB were prepared at the Institute and identified by various spectral methods. Melting points were measured on a Buchi-510 capillary apparatus. The optical rotation value [α]D was determined with Perkin-Elmer241 (589 nm). The 1H NMR spectra were recorded on Bruker AMX 400 MHz or Gemini-300 NMR spectrometer. The NMR data were reported in parts per million (ppm) relative to TMS as a reference. IR spectra were recorded on Nicolet Magna IR 750 spectrometers. The MS spectra were obtained on a Finnigan MAT 95 mass spectrometer with 70 eV sample ionization.
Pharmacology
Materials
Acetylthiocholine iodide (S-ACh), S-butyrylthiocholine iodide (S-BuCh), 5,5'-dithio-bis(2-nitrobenzoic acid) (DTNB), and tetraisopropyl pyrophosphoramide (iso-OMPA) were purchased from Sigma Chemical Company.
Enzyme source: Rat cortex homogenate was used as the brain AChE source. To prepare brain AChE, rat was decapitated and the cortex was dissected on ice and homogenized in 9 (w/v) volumes of ice-cold sodium phosphate buffer (75 mmol/L). Rat serum was used as the source of BuChE. The serum was obtained after centrifugation (3500×g, 15 min). All enzyme sources were kept at −20 °C until used.
Cholinesterase activity assay
AChE and BuChE activities were measured by the Ellman's spectrophotometric method with minor modifications22. S-ACh 2 mmol/mL or S-BuCh 2 mmol/mL were used as substrates for assays of AChE or BuChE activities, respectively. The mixture, including substrates, 1 mL sodium phosphate buffer (0.1 mmol/L), and 0.1 mL enzyme, was incubated in a total volume of 4 mL at 37 °C for 8 min. The reaction was terminated by adding 1 mL 3% SDS, and 1 mL 0.2% DTNB was then added to produce the yellow anion of 5-thio-2-nitrobenzoic acid. The rate of color production was measured spectrophotometrically at 440 nm. Enzyme activity is expressed as a percentage of the activity observed in the absence of inhibitor. The IC50 was defined as the concentration of inhibitor necessary to yield 50% inhibition of enzyme activity. The Ki value was determined using a Lineweaver-Burke plot. A plot of 1/OD versus the inverse of substrate concentration was also made so as to distinguish modes of AChE inhibition. All the samples were tested in duplicate.
Results
Chemical preparation and cholinesterase inhibitory activities of 16-substituted HupB derivatives
New preparative method has to be developed for the 16-substituted bifunctional HupB derivatives. At first, methyl group at the allylic 16 position of HupB was directly oxidized with SeO2 to produce the corresponding aldehyde, but following reductive-amination with a tether of nitrogen-containing chain gave complicated products.
Therefore, a more practical method was developed for preparing the 16-substituted derivatives. The secondary amino group in HupB was first converted with trifluoroacetic anhydride into the corresponding trifluoroacetyl amide 3, in which pyridone-2 was then protected to produce a less polar methoxy-pyridine 4 with Ag2CO3/MeI. The key intermediate aldehyde 5 was obtained by allylic oxidation of the methyl group in 4 with SeO2 (90% yield). Then, the aldehyde 5 was condensed with a diamine tether chain tied an aromatic group in the presence of NaBH4 and Ti(OiPr)4 to produce reductive-amination products23. The monosubstitued N-arylmethyl piperazines were first used as the tether to generate products 6a–e. During the reductive-amination reaction, the N-trifluoroacetyl group could be simultaneously removed by moderately extending the reaction time. Finally, the O-methyl pyridone in series 6 was deprotected with Me3SiCl/NaI to produce the 16-substituted derivatives 7a–e (Scheme 1).
Pharmacological tests of the 16-substituted derivatives were first performed in vitro via examining the inhibitory activity toward AChE. Surprisingly, the 7a–e derivatives demonstrated little inhibitory activities. Based on our experience with AChE inhibitors, a piperazine cycle as the tether might be a handicap. Possibly, the tether with the cycle was too rigid to bind well with the enzyme and therefore the inhibition was very low.
Our previous studies have indicated that the enhancement in potency varied with different tether lengths in the bifunctional HupB derivatives18, 19, 20. In order to search out more potent inhibitors, the aldehyde 5 was then condensed with various length N,N'-dimethyl-N-arylmethyl-alkylene- diamines to produce reductive-amination products 8a–j, which were then deprotected to yield the corresponding 16-substituted bifunctional HupB derivatives 9a–j (Scheme 1).
Pharmacological assays revealed the 9 series derivatives of HupB were generally more potent AChE inhibition than those of the parent HupB except derivative 9a, in which chain length of the tether was too short (n=6) to bind well the dual-site of the enzyme. Among the 9 series derivatives tested, the AChE inhibition studies indicated that the optimal length of the tether was a chain with decamethylenediamine between the 16-position of HupB and the arylmethyl group. Interestingly, the potencies of inhibition were not substantially changed with a different aromatic group in the derivatives of same tether length as shown in Table 1.
Among the 9 series derivatives of HupB examined, the AChE inhibitory activities of 9c, 9e, 9f, and 9i were 480 to 1360 times more potent than those of the parent HupB. Furthermore, the inhibition activities toward BuChE were 370 to 1560 times more potent than those of the parent HupB, with the IC50 values for BuChE inhibition in the range of 10-7mol/L. Apparently, the 16-substituted bifunctional 9 series derivatives of HupB were demonstrated to be multifunctional cholinesterase inhibitory agents in contrast with previous bis- and bifunctional derivatives of HupB18, 19, 20, which were highly selective towards AChE.
Chemical synthesis experiments
N-(trifluoroacetyl)-HupB (3)
To a solution of HupB (512 mg, 2 mmol) dissolved in CH2Cl2 (12 mL) and pyridine (1.2 mL), triluoroacetic anhydride (1.2 mL, 8.6 mmol) was added dropwise with magnetic stirring at about 0 °C. The mixture was stirred for 10 h and H2O (10 mL) was then added. The aqueous phase was further extracted with CH2Cl2 (10 mL×2). The combined organic phase was successively washed with aqueous NaHCO3, 5% CuSO4 and brine, and then dried over anhydrous Na2SO4. After evaporating the solvent, the residue was purified by silica column chromatography and eluted with AcOEt. The product (700 mg) was obtained with a 99% yield. mp 147−150 °C; [α]D20152.6 (c=0.52, CHCl3); IR (KBr) cm-1: 3390, 2960, 2891, 1697, 1655, 1605, 1433, 1229, 1196, 1142, 1105, 644; 1H NMR (300 MHz, CDCl3) δ: 7.44 (1H, d, J=9.4Hz), 6.48 (1H, d, J=9.3 Hz), 5.44 (1H, d, J=5.3 Hz), 3.7 (1H, d, J=14.7 Hz), 3.26 (1H, d, J=17.6 Hz), 2.94 (1H, dd, J=17.6 Hz, J=4.6 Hz), 2.72–2.45 (4H, m), 2.09–2.04 (1H, m), 1.70 (3H, br), 1.58 (3H, s), 1.41–1.35 (1H, m); MS (m/z): 352 (M+, 100), 337, 283, 255, 198, 186, 133, 84.
N-(trifluoroacetyl)-1-O-methyl-HupB (4)
To a mixture of N-(trifluoroacetyl)-HupB (719 mg, 2.04 mmol), Ag2CO3 (600 mg, 2.13 mmol) in CHCl3 (15 mL), and methyl iodide (4.5 mL, 72 mmol) was added. The mixture was stirred at room temperature (RT) avoiding light for 24 h. After filtration on Celite, the solvent was evaporated. The residue was purified by silica column chromatography and eluted with petroleum ether/ethyl acetate (2/1) to yield the product (735 mg) as an oil with a 98% yield. [α]D200.50 (c=0.57, CHCl3); IR (KBr) cm-1: 3422, 2960, 1698, 1605, 1480, 1331, 1198, 1142, 1107, 833; 1H NMR (300 MHz, CDCl3) δ: 7.52 (1H, d, J=8.3 Hz), 6.60 (1H, d, J=8.8 Hz), 5.47 (1H, d, J=5.3 Hz), 3. 90 (3H, s), 3.74 (1H, d, J=14.9 Hz), 3.26 (1H, d, J=17.4 Hz), 3.02 (1H, dd, J=18.2 Hz, J=5.50 Hz), 2.69–2.56 (3H, m), 2.46 (1H, br), 2.10 (1H, dd, J=12.4 Hz, J=3.70 Hz), 1.74–1.66 (3H, m), 1.56–1.55 (4H, m); MS (m/z): 366 (M+, 100), 351, 297, 269, 212, 147.
N-(trifluoroacetyl)-1-O-methyl-16-oxo-HupB (5)
N-(trifluoroacetyl)-O-methyl-HupB (735 mg, 2.0 mmol), SeO2 (356 mg, 3.2 mmol) and dioxane (30 mL) were placed in a flask. The mixture was heated to reflux with stirring for 8 h. After filtration and concentration, the residue was purified by silica gel chromatography and eluted with petroleum ether/ethyl acetate (2/1). The product (740 mg) was obtained with a 97% yield. mp 200–201 °C; [α]D20+136.2 (c=0.51, CH3OH); IR (KBr) cm-1: 3434, 2933, 1701, 1600, 1655, 1480, 1424, 1331, 1193, 1140, 1095, 834; 1H NMR (400 MHz, CDCl3) δ: 9.38 (1H, s), 7.57 (1H, d, 8.7 Hz), 6.81 (1H, dd, J=5.8 Hz, J=1.9 Hz), 6.62 (1H, d, J=8.7 Hz), 3.88 (3H, s), 3.81 (2H, d, J=18.4 Hz), 3.23 (1H, dd, J=18.4 Hz, J=6.5 Hz), 2.87 (1H, d, J=5.3Hz), 2.78–2.64 (3H, m), 2.19 (1H, td, J=8.8, 3.7 Hz), 1.88–1.73 (4H, m); MS (m/z): 380 (M+, 100), 351, 311, 283, 97, 85, 71, 57.
General procedures of reductive-amination products for preparation of N,N'-dimethyl-N-arylmethyl-N'-[16-(1-O-methyl-HupB)]-α,ω-alkylene diamine in 6 and 8 series
Under dry nitrogen, titanium (IV) isopropoxide (285 mg, 1.0 mmol) was added to an ethanol (10 mL) solution of aldehyde 5 (190 mg, 0.5 mmol) and monosubstituted N-arylmethyl piperazine or N,N'-dimethyl-N-arylmethyl-α,ω-alkylene diamine (1.0 mmol). While stirring the mixture, sodium borohydride (38 mg, 1.0 mmol) was added dropwise at RT. The reaction was monitored by thin layer chromatography (TLC) and then quenched with water. The mixture was then evaporated in vacuo and the residue was dissolved in chloroform (25 mL). The filtrate was washed with brine and then dried over anhydrous Na2SO4. The products of the 6 and 8 series were obtained by preparative TLC and eluted with a co-solvent system (CHCl3/MeOH/NH4OH=100/10/0.5). The analytical data and yields of products of the 6 and 8 series are shown in Table 2.
General procedures for preparation of N,N'-dimethyl-N-arylmethyl-N'-(16-HupB)-α,ω-alkylene diamine in 7 and 9 series
Under dry nitrogen, trimethylsilyl chloride (0.20 mL, 1.5 mmol) was added to a mixture of N,N'-dimethyl-N-arylmethyl-N'-[16-(O-methyl-HupB)]-α,ω-alkylene diamine (0.35 mmol) from the 6 or 8 series, as well as dry sodium iodide (225 mg, 1.5 mmol) in acetonitrile (8.0 mL). The mixture was heated to reflux for 10 h. Then methanol was added and the mixture was refluxed for an additional 10 h. After cooling, the solvent was evaporated and then 20 mL CH2Cl2 and 10 mL saturated NaHCO3 were added. The water phase was extracted with CH2Cl2 (20 mL×2). The combined organic phase was dried over Na2SO4, then filtered and concentrated. The products of 7 and 9 series were obtained by preparative TLC eluting with a co-solvent system (CHCl3/MeOH/NH4OH=100/12.5/1.0). The analytical data and yields of products of 7 and 9 series were shown in Table 3.
Further pharmacological evaluation of 16-substituted HupB derivatives 9c and 9i
In order to explore the potential candidates of the 16-substituted HupB derivatives further, pharmacological trials were implemented. First, the mechanism of AChE inhibition by 9c and 9i ( purities of 9c and 9i were determined by C18 reversed-phase column of HPLC on 95.98% and 97.62% individually)was studied. Derivatives 9c and 9i were incubated with AChE and the inhibition rate was determined every minute during a 8 minute incubation. The AChE activity did not exhibit a progressive decrease during the prolonged incubation with 9c and 9i (Figure 2). Similar to HupB, 9c, and 9i combined with AChE in a reversible way. This is an important prerequisite for further developing these HupB derivatives as AD drug candidates.

Time course of cortical AChE inhibition with HupB, 9c, and 9i incubation.
To further test the AChE inhibitory properties of derivatives 9c and 9i, the substrate kinetics of enzyme inhibition were examined. The reaction velocity at four different concentrations of acetylthiocholine substrate was obtained and a 1/v vs 1/[s] relationship was calculated and plotted using Lineweaver-Burk methods. According to the plotting results (Figure 3), derivatives 9c and 9i, as well as HupB, showed a linear mixed competitive type of enzymatic inhibition because the intersection of lines occurred in the second quadrant.

Substrate kinetics of AChE inhibition. Double-reciprocal plots of rat cortical AChE after incubation with 9c and 9i.
Furthermore, the neuroprotective effects of the 9c and 9i derivatives were evaluated on H2O2-induced cytotoxicity in a PC12 cell model. After pretreatment with 9c or 9i for 2 h, PC12 cells were exposed to 200 μmol/L H2O2 for 30 min and then replaced with fresh cell culture medium. After a further 24 h incubation, 10 μL MTT (5 mg/mL) was added to evaluate cell viability. The cell survival was significantly decreased following exposure to 200 μmol/L H2O2 (P<0.01) (Figure 4). However, cultures exposed to the same amount of H2O2 in the presence of 1 μmol/L 9c or 10 μmol/L 9i appeared markedly protected (P<0.01), similar to those treated with the parent HupB24.

Neuroprotective effects of 9c and 9i on H2O2-induced PC12 cell damage. 9c and 9i were added to the culture 2 h prior to H2O2 addition. The cells were incubated with fresh medium for 24 h. The cell viability was assessed by measuring the MTT reduction. Three independent experiments were carried out in triplicate. The data were means±SD. cP<0.01 vs control group, fP<0.01 vs hydrogen peroxide group.
Discussion
AD presents as a complex multifaceted disease. Thus, it is reasonable to assume that future treatments for AD will utilize multifunctional drugs with more than one therapeutic target.
In search of new rationally designed drug candidates to treat AD, many research groups have designed to develop dual binding site AChE inhibitors. These compounds are typically directed against the peripheral anionic site so as to interfere with Aβ deposition and aggregation25, 26, 27. Based on the inhibitory effect of HupB toward AChE and the structure of the AChE, we developed a novel series of 16-substituted derivatives of HupB and determined their inhibitory ability toward cholinesterase, as well as their protective effects against cytotoxic insults by hydrogen peroxide.
Our current study indicated that 9i in vitro has the highest potency among the HupB derivatives in regard to AChE inhibition. The inhibition of 9i toward AChE was 1360 times higher than that of its parent HupB, which suggested 9i effectively blocked AChE catalytic activity. Compared with USA Food and Drug Administration (FDA) and Chinese State Food and Drug Administration (SFDA) approved AChE inhibitors (donepezil, galantamine, rivastigmine and HupA) for AD treatment, the efficacy of 9i as a AChE inhibitor was more potent in vitro than galantamine, rivastigmine and HupA, and equivalent to donepezil28. Our study also indicated the inhibition by 9i was reversible since washout readily restored AChE activity. Moreover, regarding the mechanism of 9i inhibition on AChE, our Lineweaver-Burke plots indicated a mixed competitive mode, which might support the assertion that 9i possesses dual site binding properties. These findings underscore the potential therapeutic application of 9i for AD.
Furthermore, 9i was also found to inhibit BuChE effectively and was 1560 times more potent than its parent compound HupB. BuChE could also hydrolyze ACh similar to AChE and a correlation was recently found between BuChE inhibition and cognitive improvement in AD patients. In addition to a wide distribution in the serum of the human body, BuChE occurs not only in neurons and glia cells, but also in senile plaques and neurofibrillary tangles. Previous research has indicated that AChE activity decreases progressively in the brain of AD patients, while BuChE activity is preserved or even increases. This evidence indicates that BuChE might be a co-regulator for the activity of the neurotransmitter ACh. Hence, it is possible that dual inhibitors of both AChE and BuChE would represent a novel therapeutic approach for use in mild, moderate and, particularly, severe dementia29. Notably, rational approaches to generate multifunctional cholinesterase inhibitors have been reported by many research groups30, 31, 32, 33, 34.
Moreover, a number of researchers have indicated that AD is related to changes in oxidative metabolism35, 36. Compounds that possess anti-oxidative effects will also be expected to be beneficial in AD therapy. Interestingly, 9c and 9i exerted modest neuroprotection against H2O2-induced neurotoxicity.
In agreement with other research groups, we believe that the development of dual binding site AChE inhibitors, such as 9c and 9i, might be an innovative approach for the treatment of AD.
Conclusion
We have successfully developed a new preparation for novel 16-substituted bifunctional HupB derivatives, which are derived from the 16-methyl group. The novel derivatives of HupB are evidently more potent inhibitors for both AChE and BuChE than that of the parent HupB. We conclude that 9c and 9i are not only highly potent and reversible AChE inhibitors, but also exhibit a high potency toward inhibiting BuChE. In addition, they also possess neuroprotective effects against H2O2–induced insults. These encouraging findings suggest that the HupB derivatives are potential new drug candidates for AD treatment, and further exploration is needed to evaluate their pharmacological and clinical efficacies.
Author contribution
Xu-chang HE, Dong-lu BAI and Xi-can TANG designed the research experiments; Yu-fang SHI, Wei WANG, Yan FU, Yu XIA performed the research; Hai-yan ZHANG analyzed data; Hai-yan ZHANG and Xu-chang HE wrote the paper.
References
Goedert M, Spillantini MG . A century of Alzheimer's disease. Science 2006; 314: 777–81.
Youdim MB, Buccafusco JJ . Multi-functional drugs for various CNS targets in the treatment of neurodegenerative disorders. Trends Pharmacol Sci 2005; 26: 27–35.
Francis PT, Nordberg A, Arnold SE . A preclinical view of cholinesterase inhibitors in neuroprotection: do they provide more than symptomatic benefits in Alzheimer's disease? Trends Pharmacol Sci 2005; 26: 104–11.
Francotte P, Graindorge E, Boverie S, de Tullio P, Pirotte B . New trends in the design of drugs against Alzheimer's disease. Curr Med Chem 2004; 11: 1757–78.
Ji HF, Zhang HY . Multipotent natural agents to combat Alzheimer's disease. Functional spectrum and structural features. Acta Pharmacol Sin 2008; 29: 143–51.
Inestrosa NC, Alvarez A, Perez CA, Moreno RD, Vicente M, Linker C, et al. Acetylcholinesterase accelerates assembly of amyloid-beta-peptides into Alzheimer's fibrils: possible role of the peripheral site of the enzyme. Neuron 1996; 16: 881–91.
Bartolini M, Bertucci C, Cavrini V, Andrisano V . beta-Amyloid aggregation induced by human acetylcholinesterase: inhibition studies. Biochem Pharmacol 2003; 65: 407–16.
Munoz-Torrero D, Camps P . Dimeric and hybrid anti-Alzheimer drug candidates. Curr Med Chem 2006; 13: 399–422.
Castro A, Martinez A . Targeting beta-amyloid pathogenesis through acetylcholinesterase inhibitors. Curr Pharm Des 2006; 12: 4377–87.
Liu J, Zhu YL, Yu CM, Zhou YZ, Han YY, Wu FW, et al. The structure of huperzine A and B, two new alkaloids exhibiting marked anti-cholinesterase activity. Can J Chem 1986; 64: 837–9.
Bai DL, Tang XC, He XC . Huperzine A, a potential therapeutic agent for treatment of Alzheimer's disease. Curr Med Chem 2000; 7: 355–74.
Jiang H, Luo X, Bai D . Progress in clinical, pharmacological, chemical and structural biological studies of huperzine A: a drug of traditional chinese medicine origin for the treatment of Alzheimer's disease. Curr Med Chem 2003; 10: 2231–52.
Yan XF, Lu WH, Lou WJ, Tang XC . Effects of huperzine A and B on skeletal muscle and the electroencephalogram. Acta Pharmacol Sin 1987; 8: 117–23.
Zhu XD, Tang XC . Improvement of impaired memory in mice by huperzine A and huperzine B. Acta Pharmacol Sin 1988; 9: 492–7.
Xu H, Tang XC . Cholinesterase inhibition by huperzine B. Acta Pharmacol Sin 1987; 8: 18–22.
Liu J, Zhang HY, Wang LM, Tang XC . Inhibitory effects of huperzine B on cholinesterase activity in mice. Acta Pharmacol Sin 1999; 20: 141–5.
Wang ZF, Zhou J, Tang XC . Huperzine B protects rat pheochromocytoma cells against oxygen-glucose deprivation-induced injury. Acta Pharmacol Sin 2002; 23: 1193–8.
Feng S, Wang Z, He X, Zheng S, Xia Y, Jiang H, et al. Bis-huperzine B: highly potent and selective acetylcholinesterase inhibitors. J Med Chem 2005; 48: 655–7.
Feng S, Xia Y, Han D, Zheng C, He X, Tang X, et al. Synthesis and acetylcholinesterase inhibition of derivatives of huperzine B. Bioorg Med Chem Lett 2005; 15: 523–6.
He XC, Feng S, Wang ZF, Shi Y, Zheng S, Xia Y, et al. Study on dual-site inhibitors of acetylcholinesterase: Highly potent derivatives of bis- and bifunctional huperzine B. Bioorg Med Chem 2007; 15: 1394–408.
Dvir H, Jiang HL, Wong DM, Harel M, Chetrit M, He XC, et al. X-ray structures of Torpedo californica acetylcholinesterase complexed with (+)-huperzine A and (–)-huperzine B: structural evidence for an active site rearrangement. Biochemistry 2002; 41: 10810–8.
Ellman GL, Courtney KD, Andres V Jr, Feather-Stone RM . A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol 1961; 7: 88–95.
Bhattacharyya S . Reductive alkylation of dimethylamine using titanium (IV) isopropoxide and sodium borohydride: an efficient, safe and convenient method for synthesis of N,N-dimethylated tertiary amines. J Org Chem 1995; 60: 4928–9.
Zhang HY, Tang XC . Huperzine B, a novel acetylcholinesterase inhibitor, attenuates hydrogen peroxide induced injury in PC12 cells. Neurosci Lett 2000; 292: 41–4.
Bolognesi ML, Cavalli A, Valgimigli L, Bartolini M, Rosini M, Andrisano V, et al. Multi-target-directed drug design strategy: from a dual binding site acetylcholinesterase inhibitor to a trifunctional compound against Alzheimer's disease. J Med Chem 2007; 50: 6446–9.
Du DM, Carlier PR . Development of bivalent acetylcholinesterase inhibitors as potential therapeutic drugs for Alzheimer's disease. Curr Pharm Des 2004; 10: 3141–56.
Holzgrabe U, Kapkova P, Alptuzun V, Scheiber J, Kugelmann E . Targeting acetylcholinesterase to treat neurodegeneration. Expert Opin Ther Targets 2007; 11: 161–79.
Wang R, Yan H, Tang XC . Progress in studies of huperzine A, a natural cholinesterase inhibitor from Chinese herbal medicine. Acta Pharmacol Sin 2006; 27: 1–26.
Giacobini E . Cholinesterase inhibitors: new roles and therapeutic alternatives. Pharmacol Res 2004; 50: 433–40.
Savini L, Gaeta A, Fattorusso C, Catalanotti B, Campiani G, Chiasserini L, et al. Specific targeting of acetylcholinesterase and butyrylcholinesterase recognition sites. Rational design of novel, selective, and highly potent cholinesterase inhibitors. J Med Chem 2003; 46: 1–4.
Rosini M, Andrisano V, Bartolini M, Bolognesi ML, Hrelia P, Minarini A, et al. Rational approach to discover multipotent anti-Alzheimer drugs. J Med Chem 2005; 48: 360–3.
Decker M . Homobivalent quinazolinimines as novel nanomolar inhibitors of cholinesterases with dirigible selectivity toward butyrylcholinesterase. J Med Chem 2006; 49: 5411–3.
Rodriguez-Franco MI, Fernandez-Bachiller MI, Perez C, Castro A, Martinez A . Design and synthesis of N-benzylpiperidine-purine derivatives as new dual inhibitors of acetyl- and butyrylcholinesterase. Bioorg Med Chem 2005; 13: 6795–802.
Piazzi L, Belluti F, Bisi A, Gobbi S, Rizzo S, Bartolini M, et al. Cholinesterase inhibitors: SAR and enzyme inhibitory activity of 3-[omega-(benzylmethylamino)alkoxy]xanthen-9-ones. Bioorg Med Chem 2007; 15: 575–85.
Gilgun-Sherki Y, Melamed E, Offen D . Antioxidant treatment in Alzheimer's disease: current state. J Mol Neurosci 2003; 21: 1–11.
Markesbery WR . Oxidative stress hypothesis in Alzheimer's disease. Free Radic Biol Med 1997; 23: 134–47.
Acknowledgements
This project was supported by the National Natural Science Foundation of China (Grant No 30472067) and Shanghai SK Foundation for Research and Development (Project No 2004010-h).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Shi, Yf., Zhang, Hy., Wang, W. et al. Novel 16-substituted bifunctional derivatives of huperzine B: multifunctional cholinesterase inhibitors. Acta Pharmacol Sin 30, 1195–1203 (2009). https://doi.org/10.1038/aps.2009.91
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2009.91
Keywords
This article is cited by
-
Management of oxidative stress and other pathologies in Alzheimer’s disease
Archives of Toxicology (2019)
-
Retrospect and prospect of active principles from Chinese herbs in the treatment of dementia
Acta Pharmacologica Sinica (2010)
