
Abstract
Aim:
A previous study showed that individuals of Japanese descent affected by early onset familial Paget's disease of bone (PDB) carried a 27-bp duplication at position 75 (75dup27) in the TNFRSF11A gene encoding RANK. Here we report the identification of a novel mutation (78dup27) in exon 1 of TNFRSF11A in a Chinese family with early onset PDB.
Methods:
We conducted clinical and genetic studies in a non-consanguineous Chinese family with early onset PDB. The entire coding region of TNFRSF11A was amplified and directly sequenced directly.
Results:
A novel 27-bp duplication in exon 1 (78dup27) in TNFRSF11A was found in four affected individuals and one asymptomatic individual. Although this duplication was the same length as the previously identified mutation (27 bp, from bases 78 to 104), in our patients the nine duplicated amino acids in the RANK signal peptide were LLLLCALLA. The phenotypes of affected individuals in this family overlapped with both early onset PDB and classic PDB, but several distinguishing features were found in our patients. The key difference between our familial PDB and the Japanese early onset PDB was the age of onset, which in most of our patients was during their late 20s (except for the propositus' niece). Another notable difference was that the propositus' son (24 years old), who carried the 78dup27 mutation, had no clinical symptoms or bone abnormalities, except for increased serum ALP, OC and CTX.
Conclusion:
Our findings may provide a better understanding of the clinical features of early onset PDB and support the notion of a hot spot for mutations in exon 1 of the TNFRSF11A gene.
Similar content being viewed by others


Introduction
Paget's disease of bone (PDB) is a common disease characterized by focal areas of increased bone turnover, affecting one or several bones throughout the skeleton1. PDB is rare in China2. In 2000, Hughes et al3 first reported a Japanese family with early onset PDB caused by a 75dup27 mutation in the TNFRSF11A (MIM 603499) gene encoding receptor activator of nuclear factor kappa B (RANK). Early onset familial PDB is inherited as a simple autosomal dominant trait. Although the features of this disease are similar to classic PDB, several distinguishing features were found in affected subjects, including early onset and involvement of the mandible, maxilla and small bones of hands4. Furthermore, familial classic PDB is commonly caused by mutations in the sequestosome 1 (SQSTM1) gene5, 6. However, until now, TNFRSF11A mutations have not been observed in patients with the more typical form of PDB. Here, we report that a novel mutation (78dup27) in exon 1 of TNFRSF11A was identified in a Chinese family with early onset PDB. In addition, we found that these affected individuals displayed several phenotypes distinct from those of the Japanese family with early onset PDB.
Materials and methods
Patients and family
The study was approved by the Ethics Committee of the Shanghai Jiao Tong University Affiliated Sixth People's Hospital. All of the subjects, from Nanjing city in Jiangsu province in China, were recruited by our osteoporosis department and signed informed consent documents before entering the project. The pedigree of the family is shown in Figure 1. Ten members of the non-consanguineous family were investigated in the study. We diagnosed early onset PDB based on clinical symptoms of bone pain and bone deformities, radiographic features in affected bones and biochemical indices of bone turnover.

Pedigree of the family displaying early onset Paget's disease of bone. Clinical status is indicated by open symbols (unaffected) and solid symbols (affected). Proband is indicated by the arrow.
Porosities
The female propositus (III4) was born in 1959 and is now 49 years old. She was first investigated at the age of 47. She is the second of three siblings (two brothers) in the family. At 38 years old, she complained of painful expansion in knee joints as well as finger joint enlargement without pain. She did not see a doctor at that time, so the disease could not be definitively diagnosed. In 2006, she suffered from painful deformity of the lower limbs and shortening of body height. Her current body height is 150.5 cm, though at 25 years of age, her height was 158 cm. Radiographs revealed deformities of affected bones in the pelvis, vertebral spine, femora and knee joint (Figure 2A, 2B). An isotope bone scan showed increased tracer uptake in skull, clavicles, diaphysis of femora, tibias, mandible and maxilla. In addition, bent lower limbs presented a bowlegged appearance (Figure 2C). Dual energy X-ray absorptiometry (DXA) measurements showed that the bone mineral density (BMD) Z scores for the lumbar spine 1-4, femoral neck and total hip were −3.7, −0.9 and −0.4, respectively (Table 1). Serum alkaline phosphatase (ALP) was 293 IU/L. Serum calcium, phosphorus and parathyroid hormone (PTH) were in the normal range (Table 1). We gave her a preliminary diagnosis of PDB. She was treated by infusion of the bisphosphonate Ibandronate (5 mg by endovenous injection for 2 h). Three months later, her serum ALP level fell to 135 IU/L, and her pain also subsided. In August 2007, her serum ALP was 137 IU/L, and she complained of pain in the bilateral knee joints, so 5 mg of Ibandronate was again administered. Three months later, her serum ALP fell into the normal range (105 IU/L), and her BMD scores were significantly increased. Six months later (in February 2008), she was treated with the same dose of Ibandronate. In June 2008, serum ALP and PTH levels were 118 IU/L and 81.8 ng/mL, respectively. At this time, serum osteocalcin (OC) and C-terminal telopeptides of type I collagen (β-CTX) levels were analyzed by electrochemiluminescence immunoassay (ECLIA) (Roche Diagnostics GmbH, Mannheim, Germany). The intra- and interassay coefficients of variation (CV) for OC and CTX, respectively, were as follows: 6.5% and 1.6% for OC, and 4.7% and 7.6% for CTX. Serum OC and CTX levels were in the normal range as a result of Ibandronate therapy (Table 1).

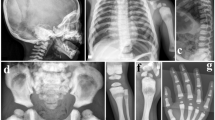
(A) Pelvis and femora radiograph of patient III4 showing osteolysis, osteosclerosis and cortical thickening. (B) Right femur radiograph of the same patient showing deformation and cortical thickening. (C) The isotope bone scan shows increased tracer uptake in affected bones.
Father
This 74-year-old (II1) man has the same condition as his daughter, but with greater severity. At the age of 30, he complained of painful enlarged knee joints, markedly bowed tibia and finger joint enlargement without pain. These afflictions severely affected his reactiveness and mobility. At that time, he presented deformity affecting the pelvis and lower limbs. At the age of 49, his left femoral neck was fractured in an accident. In 2008, he suffered a right femoral neck fracture while taking a bath. At the time of the study, he presented a significantly short stature of 133.5 cm (at age 25, his body height was 165 cm). Marked deformities were present on his head and face (Figure 3A), and his hands showed joint expansion (Figure 3B). His skull presented the classic Paget's bone phenotype (Figure 3C). Radiographs showed deformity of affected bones in the pelvis, vertebral spine, femora, and knee joints with bony expansion, osteolysis and osteosclerosis (Figure 3D, 3E). Serum ALP was 136 IU/L, and serum OC and CTX were significantly elevated at 188.3 ng/mL and 2915.0 pg/mL, respectively. Serum calcium, phosphorus and PTH were in the normal range. Other laboratory findings and BMD values are presented in Table 1.

(A) Photograph of patient II1 showing facial deformity with maxillary expansion. (B) Photograph of the hand of the same patient showing swelling of the proximal interphalangeal and distal interphalangeal joints. (C) Skull radiograph showing osteolysis and osteosclerosis. (D) and (E) Right and left femora radiographs showing deformity and bony expansion with osteolysis and osteosclerosis.
Brother
The propositus' brother (III5) was first investigated at the age of 46. At 28 years of age, he presented painfully enlarged knee joints, markedly bowed tibia and finger joint expansion (Figure 4A, 4B). At age 42, he suffered a right upper femoral fracture while walking. At that time, he was not diagnosed with PDB and was given no medical treatment. At present, he complains of deformity affecting the spine, pelvis and lower limbs. We found that his bent lower limbs presented a bowlegged appearance. His current body height and weight are 153 cm and 52 kg. At the age of 25, his body height was 170 cm. Five years ago, hearing in his left ear was affected, and he lost three teeth in the upper jaw due to inflammation. At the time of the study, radiographs showed deformity of affected bones in the skull, pelvis, vertebral spine, femora and knee joints, with bony expansion, osteolysis and osteosclerosis (Figure 4C–4E). The isotope bone scan showed increased tracer uptake in the skull, clavicles, humerus, ulna, radius, femora, tibias, mandible and maxilla (Figure 4F). Serum ALP, OC and CTX were significantly elevated (Table 1).

(A) Photograph of hands of patient III5 showing swelling of the proximal interphalangeal and distal interphalangeal joints. (B) Hands of the same patient showing expansion of phalanges. (C) Lateral radiograph of skull showing bony expansion, patchy osteolysis and osteosclerosis in the vertex and occipitalia, and bone expansion and osteosclerosis in the mandible and maxilla. (D) In right humerus, the bone is markedly expanded with osteolysis and osteosclerosis. (E) Femur radiograph of the same patient showing bone expansion and deformity with osteolysis and osteosclerosis. (F) An isotope bone scan of the same patient showing increased tracer uptake in affected bones.
Niece
The propositus' niece (IV3) is 16 years old. At 10 years old, she complained of back pain. Radiographs showed scoliosis, but no skull or hand defects were found (Figure 5). Her current body height and weight are 163 cm and 58 kg. Serum ALP was elevated (240 IU/L), and serum calcium was slightly elevated (2.79 mmol/L). These markers were measured again as follows: 187 IU/L and 174 IU/L for serum ALP and 2.66 mmol/L and 2.61 mmol/L for serum calcium. Serum phosphorus and PTH were in the normal range, while serum OC and CTX were significantly elevated (Table 1). BMD values in spine and left femur were significantly lower than those of age-matched healthy females (Table 1).

Radiograph of patient IV3 showing scoliosis.
Son
The propositus' son (IV2, 24 years old) had no clinical symptoms, and the radiographs showed no skeletal abnormalities (Figure 6A, 6B), but he displayed elevated serum ALP, OC and CTX levels (Table 1). Serum calcium, phosphorus and PTH were in the normal range. His current body height is 176 cm.

Radiograph of propositus' son (IV2) showing normal pelvis (A) and skull (B).
In addition, the propositus' grandfather (I1) was reportedly healthy and died of stroke at the age of 67. Her grandmother (I2) showed a disease onset similar to that of the propositus; at the age of 28, she presented deformity of the hand, spine and lower limbs. Her left femoral neck fractured at 70 years old, but she did not receive any medical treatment and died at the age of 73. The propositus' older brother (III1) and his daughter (IV1) were healthy.
Mutation analysis
Altogether, 110 DNA samples, including all 10 family members and 100 healthy unrelated donors of Han ethnicity, were collected. DNA was extracted from EDTA-treated peripheral blood samples using conventional methods. The DNA sequence for the TNFRSF11A gene was obtained from an online database (GeneBank accession number: AF018253). The entire coding region of TNFRSF11A was amplified with 11 pairs of primers designed using Primer 3 software (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi)(Table 2). We screened all 11 exons of TNFRSF11A using polymerase chain reaction (PCR) amplification followed by direct sequencing of the PCR fragments. We also sequenced the entire coding region of the SQSTM1 gene (GeneBank accession number: NC_000005) to screen for additional mutations. The primers used to amplify the eight SQSTM1 exons have been previously described 7.
Results
We found a 27-bp duplication of bases 78-104 (CTGCTGCTGCTCTGCGCGCTGCTCGCC) (TNFRSF11A 78dup27) in exon 1 of all affected individuals (II1, III4, III5, IV3) and one asymptomatic individual (IV2) (Figure 7A, 7B). Each person was heterozygous for the mutation. This duplication corresponds to a nine amino acid (LLLLCALLA) duplication in our patients that is distinct from the duplicated ALLLLCALL sequence observed in the affected individuals of the Japanese family with early onset PDB3, 4. The 78dup27 mutation was not identified in other members of this family (III1, III2, III3, III6, and IV1) or in 100 healthy unrelated controls. As expected, no functional mutations in SQSTM1 were found in affected individuals (data not shown).

A 27-bp duplication mutation was identified in TNFRSF11A. (A) Sequence chromograms of the sense strand for normal control. (B) Early onset PDB mutation. The 27-bp duplicated region is underlined. The region shown is from base 78 to base 104 in exon 1.
Discussion
We detected six affected members (including the deceased grandmother) in this Chinese family with early onset PDB. We identified four patients (II1, III4, III5, IV3) and one asymptomatic individual (IV2) with heterozygous mutations in TNFRSF11A. Interestingly, this novel mutation (78dup27) was different from that found by Hughes et al3. They determined that patients of an early onset PDB family of Japanese descent carried a 75dup27 mutation in TNFRSF11A. Although we also detected a 27-bp duplication in exon 1, this duplication involved bases 78–104, whereas Hughes et al reported a duplication of bases 75–101 3, 4. That is, the nine duplicated amino acids were LLLLCALLA in the Chinese patients and ALLLLCALL in affected individuals of the Japanese family3, 4.
We summarize several characteristics of this Chinese familial PDB as follows: 1. The genetic pattern is autosomal dominant; 2. The age of onset was relatively young. The propositus, her grandmother, father and brother presented symptoms at 28-38 years of age, but her niece had an age of onset of 10; 3. The disease affected the axial and skull bones as well as the small hand joints; 4. Radiography in affected bones showed osteolysis and osteosclerosis with bony expansion and deformities; 5. The isotope bone scan and radiography showed significant involvement of the mandible and maxilla in several affected individuals (II1, III4, III5); 6. Although serum ALP was mildly elevated in all patients (2–6 times above the normal range), significant elevation in serum OC and CTX levels was also observed; 7. The affected individuals had no tooth loss or deafness in childhood; 8. The propositus' son (aged 24) carried this mutation and showed elevation in serum ALP, OC and CTX, but no clinical symptoms or abnormal findings in the skeleton. Clearly, the phenotypes of affected individuals in this family overlapped with both early onset PDB and classic PDB, but several distinguishing features were found in our patients. Similar to affected individuals of the Japanese family, a particularly striking feature was found in the involvement of the mandible and maxilla in our patients (II1, III4, III5). However, a minor difference between our familial PDB and the Japanese PDB is the age of onset, which in most of our patients was during their late 20s (except for the propositus' niece), whereas in the Japanese family, onset was noted in the subjects' early 20s. Another notable feature is the propositus' son, who carried the 78dup27 mutation but had no clinical symptoms or bone abnormalities except for increased serum ALP, OC, and CTX. However, all individuals carrying the 75dup27 mutation in the Japanese familial early onset PDB had severe clinical manifestations. Therefore, we cannot presume that the minor phenotypic differences between affected subjects in this family and the Japanese family can be attributed to the shifted sequence duplication.
Indeed, exon 1 of the TNFRSF11A gene is a hot spot for mutations, which tend to present phenotypic heterogeneity8. Familial expansile osteolysis (FEO), expansile skeletal hyperphosphatasia (ESH) and the Japanese early onset PDB were caused by 84dup18, 84dup15, and 75dup27 mutations, respectively3, 4, 9, 10. In order to definitively diagnose early onset PDB, we should differentiate it from both FEO and ESH. Tooth loss and deafness in childhood are general symptoms in both familial FEO and ESH3, 4, 9, 10. Furthermore, isotope bone scans showed increased tracer uptake in the mandible and maxilla in the Chinese patients of the present study. These particular findings, combined with the identified 78dup27 mutation in our affected individuals, supported the exclusion of diagnosis for FEO and ESH.
The TNFRSF11A gene encodes RANK, which is essential for osteoclast formation. This has been demonstrated in RANK-/- mice, which lack osteoclasts and have a severe defect in bone resorption and remodeling11, 12. Hughes et al3 identified six and nine amino acid insertion mutations affecting the signal peptide region of the RANK protein, which segregated with the disease in affected individuals from FEO and early onset PDB, respectively. The 78dup27 and 75dup27 mutations may affect RANK expression and NF-κB activation in vivo, but the reason underlying their slightly different effects is unclear. Therefore, it would be of interest to know whether the 78dup27 mutation also increases cytoplasmic RANK protein levels, similar to the duplication already reported3.
Sporadic and familial PDB are rare in Japanese and Chinese individuals, so this identification of early onset familial PDB was somewhat fortuitous. Our findings, combined with the results from the cases of Japanese PDB, show that early onset familial PDB of Asian descent can be caused by a gene mutation different from that which typically underlies familial PDB in Caucasian patients. The latter is commonly caused by SQSTM1 mutations5, 6, 7. No functional mutations in TNFRSF11A were found in six Caucasian patients with early onset PDB (age of onset below 40 years) or in 64 individuals affected with familial PDB of Caucasian origin16. So far, no SQSTM1 mutations have been detected in Asian PDB patients. We presume that the different genetic backgrounds of Caucasians and Asians affect the pathogenesis of familial PDB.
This study made the unique finding that the propositus' son, who carried the 78dup27 mutation, had no clinical symptoms or abnormal features in the skeleton, which is notably different from the previously reported cases4. However, in addition to the elevation of markers of bone formation such as serum ALP and OC, all mutation carriers, including the son, displayed significantly increased serum CTX. This suggested high bone resorption activity, which can lead to bone abnormalities. Because the age of onset in most patients was 28–38 years old in the Chinese family and at present the propositus' son is only 24 years old, we conclude that his relative youth explains why no abnormal bone features were found.
In addition, in this study, the propositus was treated by infusion of the bisphosphonate Ibandronate. We found that Ibandronate therapy can significantly suppress bone turnover and relieve bone pain. Our results were similar to findings recently reported by Riches et al, and we are therefore in complete agreement with their conclusion17. Intravenous bisphosphonate therapy may be the preferred mode of treatment for early onset PDB, as it appears to provide long-term suppression of bone turnover17.
In conclusion, we identified a novel tandem duplication (78dup27) in exon 1 of the TNFRSF11A gene in an early onset PDB family of Chinese descent. The clinical features observed in affected members of this family were slightly different from those of early onset PDB of Japanese origin. Our findings provide a better understanding of the clinical features of early onset PDB and support the notion of a hot spot for mutations in exon 1 of the TNFRSF11A gene.
Author contribution
Zhen-lin ZHANG designed research; Yao-hua KE and Hua YUE performed research; Jin-wei HE, Yu-juan LIU and Yao-hua KE recruited patients; Zhen-lin ZHANG and Hua YUE wrote the paper.
References
Ralston SH, Langston AL, Reid IR . Pathogenesis and management of Paget's disease of bone. Lancet 2008; 372: 155–63.
Zhang ZL, Meng XW, Xing XP, Wang O, Xia WB, Li M, et al. Prospective study of pamidronate disodium in treatment of Paget's disease of bone. Zhonghua Yi Xue Za Zhi 2003; 83: 1653–6.
Hughes AE, Ralston SH, Marken J, Bell C, MacPherson H, Wallace RG, et al. Mutations in TNFRSF11A, affecting the signal peptide RANK, cause familial expansile osteolysis. Nat Genet 2000; 24: 45–8.
Nakatsuka K, Nishizawa Y, Ralston SH . Phenotypic characterization of early onset Paget's disease of bone caused by a 27-bp duplication in the TNFRSF11A gene. J Bone Miner Res 2003; 18: 1381–5.
Good DA, Busfield F, Fletcher BH, Lovelock PK, Duffy DL, Kesting JB, et al. Identification of SQSTM1 mutations in familial Paget's disease in Australian pedigrees. Bone 2004; 35: 277–8.
Leach RJ, Singer FR, Ench Y, Wisdom JH, Pina DS, Johnson-Pais TL . Clinical and cellular phenotypes associated with sequestosome 1 (SQSTM1) mutations. J Bone Miner Res 2006; 21 (Suppl2): P45–50.
Laurin N, Brown JP, Morissette J, Raymond V . Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in Paget disease of bone. Am J Hum Genet 2002; 70: 1582–8.
Palenzuela L, Vives-Bauza C, Fernández-Cadenas I, Meseguer A, Sarret E, Schwartz S, et al. Familial expansile osteolysis in a large Spanish kindred resulting from an insertion mutation in the TNFRSF11A gene. J Med Genet 2002; 39: E67.
Johnson-Pais TL, Singer FR, Bone HG, McMurray CT, Hansen MF, Leach RJ . Identification of a novel tandem duplication in exon 1 of the TNFRSF11A gene in two unrelated patients with familial expansile osteolysis. J Bone Miner Res 2003; 18: 376–80.
Whyte MP, Hughes AE . Expansile skeletal hyperphosphatasia is caused by a 15-base pair tandem duplication in TNFRSF11A encoding RNA and is allelic to familial expansile osteolysis. J Bone Miner Res 2002; 17: 26–9.
Li J, Sarosi I, Yan XQ, Morony S, Capparelli C, Tan HL, et al. RANK is the intrinsic hematopoietic cell surface receptor that controls osteoclastogenesis and regulation of bone mass and calcium metabolism. Proc Natl Acad Sci U S A 2000; 97: 1566–71.
Morissette J, Laurin N, Brown JP . Sequestosome 1: mutation frequencies, haplotypes, and phenotypes in familial Paget's disease of bone. J Bone Miner Res 2006; 21(Suppl2):P38–44.
Martini G, Gennari L, Merlotti D, Salvadori S, Franci MB, Campagna S, et al. Serum OPG and RANKL levels before and after intravenous bisphosphonate treatment in Paget's disease of bone. Bone 2007; 40: 457–63.
Alvarez L, Peris P, Guañabens N, Vidal S, Ros I, Pons F, et al. Serum osteoprotegerin and its ligand in Paget's disease of bone: relationship to disease activity and effect of treatment with bisphosphonates. Arthrit Rheumatism 2003; 48: 824–8.
Ralston SH . Pathogenesis of Paget's disease of bone. Bone 2008; 43: 819–25.
Sparks AB, Peterson SN, Bell C, Loftus BJ, Hocking L, Cahill DP, et al. Mutation screening of the TNFRSF11A gene encoding receptor activator of NF kappa B (RANK) in familial and sporadic Paget's disease of bone and osteosarcoma. Calcif Tissue Int 2001; 68: 151–5.
Riches PL, Imanishi Y, Nakatsuka K, Ralston SH . Clinical and biochemical response of TNFRSF11A-mediated early-onset familial Paget disease to bisphosphonate therapy. Calcif Tissue Int 2008; 83: 272–5.
Acknowledgements
This project was supported by the National Natural Science Foundation of China (No 30570891, 30771019, and 30800387) and the Program of Shanghai Subject Chief Scientist (No 08XD1403000).
We thank the patients for their participation.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ke, Yh., Yue, H., He, Jw. et al. Early onset Paget's disease of bone caused by a novel mutation (78dup27) of the TNFRSF11A gene in a Chinese family. Acta Pharmacol Sin 30, 1204–1210 (2009). https://doi.org/10.1038/aps.2009.90
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2009.90
Keywords
This article is cited by
-
Familial Paget’s disease of bone with ocular manifestations and a novel TNFRSF11A duplication variant (72dup27)
Journal of Bone and Mineral Metabolism (2023)
-
Genetic disorders associated with the RANKL/OPG/RANK pathway
Journal of Bone and Mineral Metabolism (2021)
-
Rare Inherited forms of Paget’s Disease and Related Syndromes
Calcified Tissue International (2019)
-
Paget's disease of bone—genetic and environmental factors
Nature Reviews Endocrinology (2015)
-
Exclusion of TNFRSF11B as Candidate Gene for Otosclerosis in Campania Population
Indian Journal of Otolaryngology and Head & Neck Surgery (2014)
