
Abstract
Many patients with type 2 diabetes mellitus (DM2) are at risk for micro and macro vascular complications, which could be observed in heavy smokers. Cigarette smoking increases the risk for type 2 diabetes incidence. Nicotine, acknowledged as the major pharmacologically active chemical in tobacco, is responsible for the association between cigarette smoking and development of diabetes. This minireview summarized recent studies on nicotine effects on insulin action and insulin secretion, indicating the impact of nicotine on type 2 diabetes development.
Similar content being viewed by others
Introduction
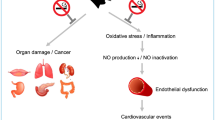
Smoking is a well-known risk factor for coronary heart disease1, 2, which is caused probably by vascular disability via its pathological change such as arteriosclerosis through the mechanisms of inflammation and endothelial dysfunctions3, 4, 5. Smoking also increases sympathetic nerve activity, which in turn increases vascular tone, increases energy expenditure, secretes corticoid6, 7, 8, and leads to heart overburden. Diabetes also causes micro- and macro vascular complications resulted from prolonged-hyperglycemia9, 10. To some extent, the effects in physical conditions of smoking and diabetes are similar, which brings a question if there is association between smoking and diabetes. After decades of studies, accumulating lines of epidemic evidence have suggested that chronic smokers have a higher risk to be insulin resistant, exhibit several aspects of the insulin resistance syndrome, and develop type 2 diabetes mellitus (DM2)11, 12, 13, 14.The risk is highly linked to the smoking degree and independent of smoking induced vascular effects12, 15.It was reported that heavy smokers (at least 20 cigarettes daily) had a 61% higher risk, while less than 20 cigarettes daily were correlated to a 29% increase of the risk12. Former smokers had just a 23% higher risk12. An appropriate temporal relationship that the cigarette smoking preceded diabetes incidence was revealed in above studies. Meanwhile, these findings are consistent with a dose-response relationship, with stronger associations for heavy smokers relative to lighter smokers and for active smokers relative to former smokers. Thus, these findings show that cigarette smoking is associated with an increased risk of type 2 diabetes and chronic smokers have a higher risk to be insulin resistant or inadequate compensatory insulin secretion responses12, 14.
To discuss the concern of smoking in DM2, one point of view is the etiology of smoking-related DM2 and another point of view is the aggravation of DM2 by smoking. Since nicotine is the critical substance which exerts most effects of smoking, we concentrate the action of nicotine on the above points in this minireview.
Nicotine on insulin action
A number of experimental and clinical studies suggest that smoking decreases insulin sensitivity, and consequently results in the disorders of glucose and lipid metabolism such as hyperglycemia and dyslipidemia including low HDL cholesterol and postprandial lipid intolerance16, 17, 18, 19. Particular in diabetic patients, it is clear that cigarette smoking worsens the metabolic control. A larger insulin dose is needed to achieve similar metabolic control in smoking patients as in non-smokers20.
In the experiments using rats, the postnatal growth of the offspring of nicotine-treated dams was enhanced by a possible result of an increase in body fat21. At the age of 26 weeks, the fasting serum insulin levels of the offspring of nicotine-treated dams were higher compared to those of control although the fasting serum glucose levels were similar in different groups of the offspring. In addition, the pattern of serum glucose levels of the offspring of nicotine-treated dams in response to an oral glucose load showed the higher numeral values at 30 min and slower recovery measured at 120 min after the glucose load, as looking like DM2, compared to those of control offspring21. According to experiments with glucose-clamp method22 to healthy volunteers and DM2 patients, nicotine infusion showed no different levels of serum insulin in healthy volunteers but required higher levels in DM2 patients23. This indicates that nicotine is more sensitive to DM2 patients in impairing insulin action of reducing blood glucose levels. This finding further suggests that nicotine given at least acutely does not directly impair insulin action but inhibits insulin mediating glucose metabolism through newly born materials and/or mechanisms appeared with DM2. Another study was undertaken to determine whether the insulin-dependent phase of postexercise muscle glycogen synthesis is impaired in a fasting population of young healthy cigarette smokers. Impaired insulin-dependent muscle glycogen synthesis was observed in these young healthy smokers, much like that observed in prediabetic subjects24.
Taken together, above findings indicate that either acute or chronic nicotine exposures could negatively affect insulin action both in smokers preceded before DM2 and DM2 patients to develop insulin resistance by affecting insulin action.
Nicotine on islet beta-cells
Nicotine influences insulin secretion through nAChRs on beta-cells Recently, many studies have found neuronal nicotinic acetylcholine receptors (nAChRs) expressed on many different non-neuronal cell types including pancreatic islet cells25, 26, 27, 28. Basal insulin secretion can be modulated by an endogenous pancreatic ganglionic mechanism. The effects of ganglionic pre- and postsynaptic nAChRs antagonism were studied in the in vitro canine pancreas. Results suggest that nAChRs are present at the ganglionic level in the pancreas and modulate insulin secretion by a complex intraganglionic mechanism29. Direct evidence of the presence of nicotinic receptors on islet beta-cells has also been obtained28. In that study, expression of mRNA for nAChRs subunits α 2, α 3, α 4, α 5, α 7, and β 2 were detected in insulin secreting cells by reverse transcriptase polymerase chain reaction. Not only a long term exposure to nicotine but also acute exposure induced a reduction in insulin secretion in response to insulin-secreating agonists including tolbutamide28. These studies suggest that nAChRs are playing an important role in controlling insulin release. Yoshikawa et al28 also has shown that acute exposure to nicotine at concentrations of higher than 1 μmol/L inhibited high-glucose-induced insulin release in isolated human islets, although these concentrations of nicotine failed to decrease the glucose-induced insulin release from rat islets. On the other hand, the exposure to nicotine for 48 h inhibited insulin release even at basal glucose levels in rat and human islets28.
These findings indicate that functional nicotinic receptors are present in pancreatic islets and beta cells and nicotine could, at least in part, negatively affect pancreatic beta-cell function. Thus, the presence of neuronal nicotinic receptors sensitive to nicotine in pancreatic cells may be a switch to modulate pancreatic cells physiological function by acetylcholine and can be involved in tobacco toxicity.
Nicotine increases apoptosis of islet β-cells There are several lines of studies have shown that nicotine can increase apoptosis of islet β-cells in nicotine exposed animal models. In a rat model, prenatal nicotine exposure affected early endocrine pancreas and adipose tissue development in pups before weaning. Results proved a direct association between fetal nicotine exposure and offspring metabolic syndrome with early signs of dysregulations of adipose tissue and pancreatic development30. Another two studies revealed that nicotine exposure can cause β-cell dysfunction, increased β-cell apoptosis, and loss of β-cell mass, which is mediated via the mitochondrial and/or death receptor pathway31, 32. This nicotine-induced apoptosis may lead to the development of postnatal dysglycemia and obesity. Recently, Bruin et al also showed that nicotine exposure caused beta-cell apoptosis and loss of β-cell mass. Moreover, this study indicated that maternally derived nicotine can act via pancreatic nAChRs during fetal and neonatal development to induce oxidative stress in the pancreas thus to induce loss of pancreatic β-cells33. These results support that beta-cell apoptosis in the fetal and neonatal pancreas can be induced by a direct effect of nicotine via nAChRs and that this effect may be mediated through increased oxidative stress33.
All these studies in animal models have indicated that prenatal or neonatal exposed to nicotine will lead to loss of pancreatic β-cells, thus less insulin secretion. Mitochondrial dysfunction, oxidative stress, and inflammation are involved as underlying mechanisms for the direct toxicity induced by nicotine via nAChRs. These findings facilitate the understanding of nicotine effects on both smoking preceding DM2 development and the aggravation of DM2.
Conclusion
Numerous clinical and experimental reports have described the association between the active cigarette smoking, the development of diabetes, glycemic control, and diabetic complications. Although confounding social factors are sometimes argued rather than a direct effect of nicotine or other cigarette components, it is probable that the most of these effects are exerted by nicotine since both prenatal and postnatal exposure to nicotine can directly induce imbalance of metabolic control. Effects of nicotine on insulin action, secretion, and development of diabetes have been widely studied both in animal models and human subjects. Results have indicated nicotine exposure could induce a reduction of insulin release, and negatively affect insulin action, suggesting nicotine could be a cause for development of insulin resistance. Mitochondrial dysfunction, oxidative stress, and inflammation are involved as underlying mechanisms of nicotine induced pancreatic β-cells loss. These evidences together indicate that cigarette smoking can affect insulin action and pancreatic cell function. To facilitate the development of diabetes, besides the well-known risk for cardiovascular and other disease. Thus, in diabetes care, smoking cessation is important for glycemic control and limiting the development of diabetic complications.
References
Burke AP, Farb A, Malcom GT, Liang Y, Smialek J, Virmani R . Coronary risk factors and plaque morphology in men with coronary disease who died suddenly. N Engl J Med 1997; 336: 1276–82.
Greenland P, Knoll MD, Stamler J, Neaton JD, Dyer AR, Garside DB, et al. Major risk factors as antecedents of fatal and nonfatal coronary heart disease events. JAMA 2003; 290: 891–7.
Cooke JP . Angiogenesis and the role of the endothelial nicotinic acetylcholine receptor. Life Sci 2007; 80: 2347–51.
Madamanchi NR, Vendrov A, Runge MS . Oxidative stress and vascular disease. Arterioscler Thromb Vasc Biol 2005; 25: 29–38.
Terry ML, Berkowitz HD, Kerstein MD. Tobacco . Its impact on vascular disease. Surg Clin North Am 1998; 78: 409–29.
Bloomgarden ZT . Obesity and diabetes. Diabetes Care 2000; 23: 1584–90.
Chrousos GP . Stressors, stress, and neuroendocrine integration of the adaptive response. The 1997 Hans Selye Memorial Lecture. Ann N Y Acad Sci 1998; 851: 311–35.
Greenway SC . Hormones in human metabolism and disease. Functional metabolism: regulation and adaptation. Weinheim: Wiley-VCH; 2004. p271.
Laakso M . Hyperglycemia as a risk factor for cardiovascular disease in type 2 diabetes. Prim Care 1999; 26: 829–39.
Schalkwijk C, Stehouwer C . Vascular complications in diabetes mellitus: the role of endothelial dysfunction. Clin Sci 2005; 109: 143–59.
Carlsson S, Midthjell K, Grill V . Smoking is associated with an increased risk of type 2 diabetes but a decreased risk of autoimmune diabetes in adults: an 11-year follow-up of incidence of diabetes in the Nord-Trondelag study. Diabetologia 2004; 47: 1953–6.
Willi C, Bodenmann P, Ghali WA, Faris PD, Cornuz J . Active smoking and the risk of type 2 diabetes: a systematic review and meta-analysis. JAMA 2007; 298: 2654–64.
Eliasson B . Cigarette smoking and diabetes. Prog Cardiovasc Dis 2003; 45: 405–13.
Facchini FS, Hollenbeck CB, Jeppesen J, Chen YD, Reaven GM . Insulin resistance and cigarette smoking. Lancet 1992; 339: 1128–30.
Mitchell BD, Hawthorne VM, Vinik AI . Cigarette smoking and neuropathy in diabetic patients. Diabetes Care 1990; 13: 434–7.
Borggreve SE, De Vries R, Dullaart RP . Alterations in high-density lipoprotein metabolism and reverse cholesterol transport in insulin resistance and type 2 diabetes mellitus: role of lipolytic enzymes, lecithin:cholesterol acyltransferase and lipid transfer proteins. Eur J Clin Invest 2003; 33: 1051–69.
Despre JP, Lemieux I . Abdominal obesity and metabolic syndrome. Nature 2006; 444: 881–7.
Heine RJ, Dekker JM . Beyond postprandial hyperglycaemia: metabolic factors associated with cardiovascular disease. Diabetologia 2002; 45: 461–75.
Smith U, Axelsen M, Carvalho E, Eliasson B, Jansson PA, Wesslau C . Insulin signaling and action in fat cells: associations with insulin resistance and type 2 diabetes. Ann N Y Acad Sci 1999; 892: 119–26.
Madsbad S, McNair P, Christensen MS, Christiansen C, Faber OK, Binder C, et al. Influence of smoking on insulin requirement and metbolic status in diabetes mellitus. Diabetes Care 1980; 3: 41–3.
Holloway AC, Lim GE, Petrik JJ, Foster WG, Morrison KM, Gerstein HC . Fetal and neonatal exposure to nicotine in Wistar rats results in increased beta cell apoptosis at birth and postnatal endocrine and metabolic changes associated with type 2 diabetes. Diabetologia 2005; 48: 2661–6.
DeFronzo RA, Tobin JD, Andres R . Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol 1979; 237: E214–23.
Axelsson T, Jansson PA, Smith U, Eliasson B . Nicotine infusion acutely impairs insulin sensitivity in type 2 diabetic patients but not in healthy subjects. J Intern Med 2001; 249: 539–44.
Price TB, Krishnan-Sarin S, Rothman DL . Smoking impairs muscle recovery from exercise. Am J Physiol Endocrinol Metab 2003; 285 : E116–22.
Conti-Fine BM, Navaneetham D, Lei S, Maus AD . Neuronal nicotinic receptors in non-neuronal cells: new mediators of tobacco toxicity? Eur J Pharmacol 2000; 393: 279–94.
Ejiri K, Taniguchi H, Baba S . Participation of nicotinic receptor in hormone release from isolated rat islets of Langerhans. Diabetes Res Clin Pract 1989; 6: 53–9.
Nelson L, Gardner ME, Young MJ . Regulation of calcium distribution in bovine sperm cells: cytochemical evidence for motility control mechanisms. Cell Motil 1982; 2: 225–42.
Yoshikawa H, Hellstrom-Lindahl E, Grill V . Evidence for functional nicotinic receptors on pancreatic beta cells. Metabolism 2005; 54: 247–54.
Stagner JI . Modulation of insulin secretion by pancreatic gang lionic nicotinic receptors. Diabetes 1986; 35: 849–54.
Somm E, Schwitzgebel VM, Vauthay DM, Camm EJ, Chen CY, Giacobino JP, et al. Prenatal nicotine exposure alters early pancreatic islet and adipose tissue development with consequences on the control of body weight and glucose metabolism later in life. Endocrinology 2008; 149: 6289–99.
Bruin JE, Gerstein HC, Morrison KM, Holloway AC . Increased pancreatic beta-cell apoptosis following fetal and neonatal exposure to nicotine is mediated via the mitochondria. Toxicol Sci 2008; 103: 362–70.
Bruin JE, Petre MA, Raha S, Morrison KM, Gerstein HC, Holloway AC . Fetal and neonatal nicotine exposure in Wistar rats causes progressive pancreatic mitochondrial damage and beta cell dysfunction. PLoS ONE 2008; 3: e3371.
Bruin JE, Petre MA, Lehman MA, Raha S, Gerstein HC, Morrison KM, et al. Maternal nicotine exposure increases oxidative stress in the offspring. Free Radic Biol Med 2008; 44: 1919–25.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Xie, Xt., Liu, Q., Wu, J. et al. Impact of cigarette smoking in type 2 diabetes development. Acta Pharmacol Sin 30, 784–787 (2009). https://doi.org/10.1038/aps.2009.49
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2009.49
Keywords
This article is cited by
-
GSTT1 and GSTM1 polymorphisms with human papillomavirus infection in women from southern Brazil: a case–control study
Molecular Biology Reports (2022)
-
Fermented pork fat (Sa-um) and lifestyle risk factors as potential indicators for type 2 diabetes among the Mizo population, Northeast India
Journal of Health, Population and Nutrition (2021)
-
Phenotypic comparison between smoking and non-smoking chronic obstructive pulmonary disease
Respiratory Research (2020)
-
Study of the magnitude of diabetes and its associated risk factors among the tuberculosis patients of Morang, Eastern Nepal
BMC Public Health (2019)
-
Health Effects of Trace Metals in Electronic Cigarette Aerosols—a Systematic Review
Biological Trace Element Research (2019)
