
Abstract
Aim:
The aim of this study was to investigate the effect of the squamosamide derivative FLZ (N-2-(4-hydroxy-phenyl)-ethyl-2-(2,5-dimethoxy-phenyl)-3-(3-methoxy-4-hydroxy-phenyl)-acrylamide) on lipopolysaccharide (LPS)-induced inflammatory mediator production and the underlying mechanism in RAW264.7 macrophages.
Methods:
RAW264.7 cells were preincubated with non-toxic concentrations of compound FLZ (1, 5, and 10 μmol/L) for 30 min and then stimulated with 10 μg/L LPS. The production of nitric oxide (NO), the expression of inducible nitric oxide synthase (iNOS) and cyclooxygenase 2 (COX-2), and the activation of nuclear factor kappa-B (NF-κB) and mitogen-activated protein kinase (MAPK) pathways were examined.
Results:
FLZ significantly inhibited the LPS-induced production of NO, as well as the expression of iNOS and COX-2 at both the RNA and the protein levels in RAW264.7 cells. The LPS-induced increase in the DNA binding activity of NF-κB and activator protein 1 (AP-1), the nuclear translocation of NF-κB p65, the degradation of the inhibitory κBα protein (IκBα) and the phosphorylation of IκBα, IκB kinase (IKK) α/β, c-Jun NH2-terminal kinase (JNK) and p38 MAPKs were all suppressed by FLZ. However, the phosphorylation of extracellular signal-regulated kinase (ERK) was not affected. Further study revealed that FLZ inhibited the phosphorylation of transforming growth factor-β (TGF-β)-activated kinase 1 (TAK1), which is an upstream signaling molecule required for IKKα/β, JNK and p38 activation.
Conclusion:
FLZ inhibited the LPS-induced production of inflammatory mediators at least partly through the downregulation of the TAK-IKK and TAK-JNK/p38MAPK pathways.
Similar content being viewed by others

Introduction
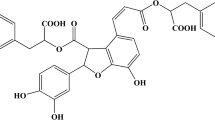
Compound FLZ (N-2-(4-hydroxy-phenyl)-ethyl-2-(2,5-dimethoxy-phenyl)-3-(3-methoxy-4-hydroxy-phenyl)-acrylamide) is a novel synthetic derivative of squamosamide (Figure 1) 1. Previous studies have demonstrated that FLZ has strong antioxidative, antiapoptotic and neuroprotective effects. FLZ has been shown to protect against mitochondrial damage and apoptosis of neurons exposed to various neurotoxins, including 6-hydroxydopamine 2, 1-methyl, 4-phenyl-pyridinium 3 and β-amyloid 4. Remarkably, FLZ has also been shown to improve experimental learning and memory deficits 5 and dyskinesia in animal models 6. Therefore, FLZ has potential as a new therapeutic compound for the treatment and/or prevention of neurodegenerative diseases.

The chemical structure of FLZ.
Inappropriate immune and inflammatory responses have been proposed to be involved in the pathogenesis of neurodegenerative diseases such as Alzheimer's disease and Parkinson's disease 7, 8, 9. The microglial cell is the resident macrophage in the brain and its overactivation may result in progressive neurotoxicity 10, 11, 12. Therefore, inhibition of the inflammatory process may be a target for the development of therapies for neurodegenerative diseases. In a previous study, we demonstrated that FLZ protected dopaminergic neurons against lipopolysaccharide (LPS)-induced neurotoxicity and the neuroprotective effect of FLZ was attributed to a reduction in LPS-induced proinflammatory factors in microglia 13. The aim of this study was to further explore the anti-inflammatory effects of FLZ and to elucidate the mechanism by which FLZ modulates inflammation.
The nuclear factor kappa-B (NF-κB) and mitogen-activated protein kinase (MAPK) pathways are the most important networks for the regulation of many genes involved in inflammatory response 14, 15. NF-κB exists mainly as a heterodimer composed of subunits of the Rel family, p50 and p65. In the resting state, NF-κB normally localizes to the cytoplasm, where it is bound by the inhibitory IκB proteins (IκBα, IκBβ, IκBɛ). However, following inflammatory stimuli, such as LPS, IκB is phosphorylated by IκB kinase (IKK) and is subsequently degraded by the proteasome. NF-κB is then released and translocates to the nucleus, where it triggers the transcription of multiple genes through the cis-acting κB element. The MAPKs are a family of serine/threonine protein kinases, including c-Jun NH2-terminal kinase (JNK) and p38 MAPK, that play important roles in inflammatory responses 15. Transforming growth factor-β (TGF-β)-activated kinase 1 (TAK1) was first identified as a kinase involved in TGF-β signaling and was later shown to be a pivotal factor for MAPK and NF-κB activation in response to TLR, IL-1R and TNFR stimulation 16. The phosphorylation of TAK1 is a common signal for the activation of downstream targets such as IKK, JNK and p38 MAPK 17, 18.
In the present study, RAW264.7 cells, a well-defined murine macrophage cell line commonly used for the study of inflammation, were used to determine the molecular mechanism underlying the inhibitory effect of FLZ on the LPS-induced inflammatory response. The effect of FLZ on the activation of the NF-κB and MAPK pathways was examined.
Materials and methods
Compound FLZ with 99.5% purity was provided by Professor Ping XIE from the Department of Pharmaceutical Chemistry, Institute of Materia Medica, Chinese Academy of Medical Science. The stock solution of FLZ was prepared in dimethyl sulfoxide (DMSO) and diluted with culture medium before use. In all experiments, DMSO was used as a vehicle control.
LPS (from Escherichia coli 055:B5), 3-(4,5)-dimethylthiazol-2-y1)-3,5-diphenyltetrazolium bromide (MTT) and sodium nitroprusside (SNP) were obtained from Sigma-Aldrich. Fetal bovine serum (FBS) was purchased from Hyclone. The electrophoretic mobility shift assay (EMSA) kit was purchased from Pierce. Double-stranded NF-κB and AP-1 oligonucleotides were synthesized by IDT (Broad St, Newark, NJ, USA). Trizol was purchased from Invitrogen. The RT-PCR kit was purchased from Promega. The nondenaturing lysis buffer, the nuclear-cytosol extraction kit and the Super ECL Plus Detection Reagent were purchased from Applygen Technologies. The antibody against inducible nitric oxide synthases (iNOS) was purchased from Upstate Biotechnology. The antibodies against cyclooxygenase 2 (COX-2), NF-κB p65, β-actin, IκBα, p-IκBα, p38, and p-p38 MAPK were obtained from Santa Cruz Biotech. The antibodies against p-IKKα/β, p-JNK, p-ERK, and p-TAK1 as well as IKKα, JNK, ERK, and TAK1 were purchased from Cell Signaling Technology.
Cell culture and treatment
The RAW264.7 cell line, a macrophage cell line, was purchased from the Cell Culture Center at the Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences. The cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin and 100 μg/mL streptomycin at 37 °C in a 5% CO2 incubator.
Cell viability
Cell viability was evaluated using the MTT assay 19. After pre-incubation for 24 h in a 96-well plate, RAW264.7 cells were treated with various concentrations of FLZ (1, 5 or 10 μmol/L) for 48 h and then the cells were incubated with 0.5 mg/mL MTT for 4 h at 37 °C. The culture medium was removed, and the cells were lysed with 200 μL DMSO and shaken for 10 min. The optical density (OD) values at 570 nm were measured using a microplate reader. The OD values in the vehicle control group were set to 100% cell viability.
Nitric oxide (NO) determination
After preincubation for 24 h in 96-well plates, RAW264.7 cells were treated with various concentrations of FLZ for 30 min and then stimulated with 10 μg/L LPS for 36 h. The production of NO was determined by assaying the concentration of nitrite in the culture supernatant 20. Briefly, 100 μL of culture supernatant was incubated with 100 μL Griess reagent (a 1:1 mixture of 1% sulfanilamide in 5% H3PO4 and 0.1% N-(1-naphthyl) ethylenediamine in distilled water) at room temperature for 20 min. Sodium nitrite was used to generate a standard curve. The OD value of the samples at 550 nm was measured.
SNP spontaneously generates NO in aqueous solution at a physiological pH. This NO can be oxidized quickly to produce a nitrite ion that can be detected with the Griess reagent 21. The SNP powder was dissolved in phosphate-buffered saline (PBS), pH 7.4, to a concentration of 100 mmol/L. The FLZ (10 μmol/L) and SNP (10 mmol/L) were prepared and incubated at 25 °C in PBS. At the indicated times, the samples were removed and the concentration of nitrite was detected using the Griess reagent.
Western blot analysis
RAW264.7 cells were pre-treated with various concentrations of compound FLZ for 30 min and then stimulated with 10 ng/mL LPS for the indicated time. Whole-cell lysates (for the determination of iNOS, COX-2, IκBα, IKKα/β, JNK, ERK, p38 MAPK and TAK1 expression levels) and nuclear extracts (for the NF-κB p65 assay) were denatured and separated by 10% SDS-polyacrylamide gel electrophoresis and electrotransferred to polyvinylidene difluoride membranes (Millipore). The membranes were blocked in a Tris-buffered saline with 0.05% Tween-20 (TBST) solution containing 5% nonfat dried milk for 2 h and then incubated overnight at 4°C with specific antibodies against iNOS, COX-2, NF-κB p65, IκBα, p-IκBα, IKKα/β, p-IKKα/β, JNK, p-JNK, ERK, p-ERK, p38, p-p38, TAK1, or p-TAK1. The expression of β-actin in the total cell lysates and histone H3 in the nuclear extracts was used to show equal protein loading. After being washed with TBST, the membranes were incubated with horseradish peroxidase-conjugated anti-rabbit or anti-mouse IgG for 2 h at room temperature. After being extensively washed with TBST, the protein bands were detected using enhanced chemiluminescence (ECL) reagent and visualized and photographed using a LAS 3000 chemiluminescence system (Fujifilm, Tokyo, Japan). All Western blot analyses were carried out at least three times. Results are expressed as the relative ratio of the specific band compared with the internal reference.
Electrophoretic mobility shift assay (EMSA)
After pretreatment with 10 μmol/L FLZ and stimulation with 10 μg/L LPS for 1 h, the ability of nuclear extracts of RAW264.7 cells to bind probes encoding the NF-κB or AP-1 consensus binding sites was determined. Nuclear extracts were prepared according to the methods of Schreiber et al 22 with a slight modification. Briefly, cells were washed with cold PBS, scraped and collected. After centrifugation at 200×g for 5 min at 4 °C, the cell pellet was resuspended in 150 μL cold buffer A (10 mmol/L HEPES, pH 7.9, 10 mmol/L KCl, 0.1 mmol/L EDTA, 0.1 mmol/L EGTA, 1 mmol/L DTT, 0.5 mmol/L PMSF, 2.5 g/L aprotinin) and allowed to swell on ice for 30 min. Then, 9 μL of 10% Nonidet P-40 was added and the tubes were vortexed for 30 s. After incubation for 1 min on ice, the cells were centrifuged at 200×g for 5 min at 4 °C. The pellet was resuspended in 30 μL cold buffer B (20 mmol/L HEPES, pH 7.9, 0.4 mol/L NaCl, 1 mmol/L EDTA, 1 mmol/L EGTA, 1 mmol/L DTT, 1 mmol/L PMSF, 2.5 g/L aprotinin) and the tubes were incubated for 30 min at 4 °C with vortexing at 5 min intervals. The nuclear extracts were then centrifuged at 12 000×g for 15 min at 4 °C and the supernatant was frozen in aliquots at −70 °C.
Double-stranded DNA probes containing the consensus binding site of NF-κB (5′ biotin-GCCTGGGAAAGTCCCCTCAACT-3′) and AP-1 (5′biotin-CGCTTGATGACTCAGCCGGAA-3′) were used for gel shift analyses. The reaction mixture consisted of a 20 μL total volume containing 10 μg protein extract, 20 fmol biotin-labeled DNA, 2.5% glycerol, 5 mmol/L MgCl2 and 50 mg/L poly (dI-dC). The mixtures were incubated for 20 min at room temperature. DNA-protein complexes were resolved by electrophoresis in 6% polyacrylamide gels at 4 °C in 0.5×TBE buffer (45 mmol/L Tris borate, 1 mmol/L EDTA) and transferred to a nylon membrane. The membrane was then UV-cross-linked using a hand-held UV lamp equipped with a 254 nm bulb. The membrane was blocked in blocking buffer for 15 min and incubated in the conjugate/blocking buffer solution for 15 min. The membrane was then washed four times with washing buffer. The membrane was incubated in the substrate working solution (including luminal/enhancer solution and stable peroxide solution) for 5 min and bands were detected and visualized using a LAS 3000 chemiluminescence system (Fujifilm, Tokyo, Japan).
Reverse transcription-polymerase chain reaction (RT-PCR)
RAW264.7 cells were pre-treated with compound FLZ for 30 min and then stimulated with 10 μg/L LPS for 8 h. Total RNA was isolated using the Trizol reagent according to the manufacturer's protocol. RT-PCR was performed using a One-Step RT-PCR Kit. Each reaction contained 10 μL AMV/Tfl reaction buffer, 0.2 mmol/L dNTP, 1 μmol/L of each primer, 1 mmol/L MgSO4, 0.1 U/μL AMV reverse transcriptase, Tfl DNA polymerase and approximately 1 μg RNA. RT-PCR was performed in a PTC-200 Peltier thermal cycler (MJ Research, USA). The reaction was heated to 45 °C for 45 min for reverse transcription and 94 °C for 2 min for AMV RT inactivation and RNA/cDNA/primer denaturation. Then, 40 cycles of PCR were performed for iNOS and COX-2 and 25 cycles for β-actin. The denaturation, annealing and extension steps for iNOS and COX-2 amplification were 94 °C for 30 s, 58 °C for 1 min, and 68 °C for 2 min. The final extension was at 68°C for 7 min. The following primers were used in the PCR reactions and were synthesized by Shanghai Sangon Biological Engineering Technology & Services Company, China: iNOS, forward: 5′-TCAGATCCCGAAACGC-3 and reverse: 5′-CCACAACTCGCTCCAAGA-3′, which amplified a 302 bp product; COX-2, forward: 5′-CAGCAAATCCTTGCTGTTCC-3′ and reverse: 5′-TGGGCAAAGAATGCAAACATC-3′, which amplified a 517 bp product; and β-actin, forward: 5′-TGGAATCCTGTGGCATCCATGAAAC-3′ and reverse: 5′-TAAAACGCAGCTCAGTAACAGTCCG-3′, which produced a 349 bp product. The PCR products were separated by electrophoresis in a 1% agarose gel and stained with ethidium bromide. Bands were visualized and photographed using a Kodak Gel Logic 100 Imaging system (Life Technologies, Inc., Eastman Kodak, New Haven, CT) and the band densities were quantified using Gel-Pro Analyzer4.0 software. The results were normalized to the expression of the β-actin housekeeping gene.
Statistical analysis
Results are expressed as means±SD. Data were analyzed using a one-way analysis of variance (ANOVA). Values of P<0.05 were considered statistically significant.
Results
Cytotoxicity of FLZ
For the following experiments, the initial step was to determine the effect of FLZ on the viability of RAW264.7 cells. The results of the MTT assay indicated that after exposure to 1, 5, and 10 μmol/L FLZ for 48 h, the viability of RAW264.7 cells was essentially the same as that of untreated controls (Table 1), demonstrating that FLZ at concentrations of 1, 5, and 10 μmol/L is safe. Thus, 1, 5, and 10 μmol/L of FLZ was used throughout the study.
FLZ inhibits NO production in LPS-stimulated RAW264.7 cells
The concentration of nitrite was measured as an index of NO production. As shown in Figure 2A, the concentration of nitrite in the supernatant of resting cells was very low. However, after stimulation with 10 μg/L LPS for 36 h, the nitrite production increased 10-fold compared with that of the control cells. Pre-incubation of RAW264.7 cells with various concentrations of FLZ suppressed the LPS-induced increase of nitrite. FLZ alone (10 μmol/L) showed no effect on the production of NO by RAW264.7 cells.

Effect of FLZ on NO production. (A) RAW264.7 cells were cultured with 10 μg/L LPS in the presence or absence of FLZ for 36 h. The nitrite in supernatants was determined. (n=4). cP<0.01 compared with the normal group; fP<0.01 compared with the normal LPS-treated group. (B) FLZ (10 μmol/L) and sodium nitroprusside (10 mmol/L in PBS) were incubated at 25 °C for 180 min. At 30 min intervals, samples of the reaction were removed and the nitrite was detected. n=4. Data are presented as the mean±SD.
SNP (sodium nitroprusside) spontaneously generates nitrite in PBS at 25 °C. In contrast, 10 μmol/L FLZ had no effect on the generation of nitrite from SNP (Figure 2B).
FLZ inhibits LPS-induced iNOS and COX-2 protein expression in RAW264.7 cells
As detected by Western blot, the expression of the iNOS and COX-2 proteins in resting RAW264.7 cells was minimal. However, following stimulation with LPS for 24 h, the levels of iNOS and COX-2 protein significantly increased. All concentrations of FLZ tested (1, 5, and 10 μmol/L) attenuated the LPS-induced increase in the expression of both iNOS (Figure 3A) and COX-2 (Figure 4A).

Effect of FLZ on LPS-induced iNOS protein and mRNA expression in RAW264.7 cells. (A) iNOS protein was detected by Western blot. RAW264.7 cells were pretreated with various concentrations (1, 5, and 10 μmol/L) of FLZ for 30 min; then LPS (10 μg/L) was added and the cells were incubated for 24 h. Total cellular proteins (40 μg) were subjected to Western blot analysis using an antibody against iNOS. (B) RT-PCR detection of iNOS mRNA. RAW264.7 cells were pretreated with different concentrations (1, 5, and 10 μmol/L) of FLZ for 30 min; then LPS (10 μg/L) was added and the cells were incubated for 8 h. iNOS mRNA was detected with RT-PCR and agarose gel electrophoresis as described under Methods. n=3. Values are presented as the mean±SD. cP<0.01 compared with the normal group; fP<0.01 compared with the LPS-treated group.

Effect of FLZ on LPS-induced COX-2 protein and mRNA expression in RAW264.7 cells. (A) COX-2 protein expression was detected by Western blot. The cells were treated with various concentrations (1, 5, and 10 μmol/L) of FLZ for 30 min; then LPS (10 μg/L) was added and the cells were incubated for 24 h. Cellular proteins (40 μg) were subjected to Western blot analysis using an antibody against COX-2. Experiments were repeated three times. (B) COX-2 mRNA detection. RAW264.7 cells were pretreated with various concentrations (1, 5, and 10 μmol/L) of FLZ for 30 min; then LPS (10 μg/L) was added and the cells were incubated for 8 h. COX-2 mRNA was detected by RT-PCR and agarose gel electrophoresis as described under Methods. n=3. Values are presented as the mean±SD. cP<0.01 compared with the normal group; eP<0.05; fP<0.01 compared with the LPS-treated group.
FLZ inhibits LPS-induced iNOS and COX-2 mRNA expression in RAW264.7 cells
RT-PCR demonstrated that the iNOS and COX-2 mRNA expression dramatically increased after stimulation with LPS for 8 h compared with unstimulated cells (Figure 3B, Figure 4B). Additionally, the pre-incubation of RAW264.7 cells with various concentrations of FLZ inhibited the LPS-stimulated increase in iNOS and COX-2 mRNA.
FLZ inhibits LPS-induced NF-κB and AP-1 activation in RAW264.7 cells
NF-κB and AP-1 activation have been shown to be the key mechanisms underlying the LPS-induced increase in iNOS and COX-2 expression. Therefore, the binding activity of NF-κB and AP-1 in nuclear extracts from RAW264.7 cells pretreated with 10 μmol/L FLZ and stimulated with 10 μg/L LPS for 1 h was investigated. As shown in Figure 5A, the LPS-induced increase in the DNA binding of NF-κB and AP-1 was suppressed by the addition of 10 μmol/L FLZ.

Effects of FLZ on the LPS-induced DNA binding activity of NF-κB and AP-1, nuclear translocation of NF-κB p65 and degradation of IκBα in RAW264.7 cells. (A) Cells were treated with the indicated concentrations of FLZ for 30 min before stimulation with LPS for 1 h. The nuclear proteins were extracted and tested for DNA binding to NF-κB or AP-1 consensus binding sequences by EMSA. (B) NF-κB p65 assay. The cells were treated with 1, 5, or 10 μmol/L of FLZ for 30 min and then with LPS (10 μg/L) for 1 h. Nuclear proteins (40 μg) were analyzed by Western blot. (C) IκB assay. RAW264.7 cells were treated with 1, 5, or 10 μmol/L of FLZ for 30 min and then with LPS 10 μg/L for 30 min. Total cellular proteins (40 μg) were analyzed by Western blot using an antibody against IκBα. n=3. Values are presented as the mean±SD. cP<0.01 compared with the normal group; fP<0.01 compared with the LPS-treated group.
FLZ inhibits LPS-induced NF-κB p65 nuclear translocation and IκBα degradation in RAW264.7 cells
The levels of NF-κB p65 in the nuclear extracts of LPS-stimulated RAW264.7 cells were analyzed via Western blot (Figure 5B). The level of NF-κB p65 in the nucleus significantly increased following stimulation with LPS for 1 h. FLZ inhibited the LPS-induced nuclear translocation of NF-κB p65 in RAW264.7 cells. Furthermore, IκBα was dramatically degraded 30 min after exposure to 10 μg/L LPS. FLZ also significantly inhibited the LPS-induced IκBα degradation (Figure 5C).
FLZ inhibits the LPS-induced phosphorylation of IκBα and IKKα/β in RAW264.7 cells
RAW264.7 cells were preincubated for 30 min with various concentrations of FLZ and then stimulated with 10 μg/L LPS. Five minutes after stimulation with LPS, the levels of p-IκBα and p-IKKα/β were significantly increased in untreated cells. However, pretreatment with various concentrations of FLZ inhibited the increase in the phosphorylation of IκBα and IKKα/β (Figure 6A, 6B).

Effect of FLZ on the LPS-induced phosphorylation of IκBα and IKKα/β in RAW264.7 cells. RAW264.7 cells were treated with FLZ for 30 min and then with LPS (10 μg/L) for 5 min. Total cellular proteins (40 μg) were subjected to Western blot analysis using an antibody against p-IκBα (A) and p-IKKα/β (B). n=3. Values are presented as the mean±SD. bP<0.05, cP<0.01 compared with the normal group; eP<0.05, fP<0.01 compared with the LPS treated group.
FLZ inhibits the LPS-induced activation of MAPKs in RAW264.7 cells
The MAPK pathway has been implicated in the LPS-induced production of iNOS and COX-2 23, 24. Following stimulation with 10 ng/mL LPS for 20 min, the phosphorylation of JNK, ERK and p38 MAPK increased rapidly in RAW264.7 cells. However, preincubation with various concentrations of FLZ inhibited the phosphorylation of JNK and p38 MAPK, whereas the phosphorylation of ERK was not affected (Figure 7).

Effect of FLZ on the LPS-induced phosphorylation of ERK, JNK, and p38 MAPK in RAW264.7 cells. RAW264.7 cells were treated with FLZ for 30 min and then with LPS (10 ng/mL) for 20 min. Total cellular proteins (40 μg) were subjected to Western blot analysis using the indicated antibody.
FLZ inhibits the LPS-induced phosphorylation of TAK1 in RAW264.7 cells
Because TAK1 is the upstream signaling molecule of IKK, JNK and p38 MAPK, the effect of FLZ on the LPS-induced phosphorylation of TAK1 was investigated. Following a 5-min stimulation of RAW264.7 cells with LPS, an increase in p-TAK1 expression was detected by Western blot. However, preincubation with FLZ inhibited this increase in TAK1 phosphorylation (Figure 8).

Effect of FLZ on the LPS-induced phosphorylation of TAK1 in RAW264.7 cells. RAW264.7 cells were treated with FLZ for 30 min and then with LPS (10 μg/L) for 5 min. Total cellular proteins (40 μg) were subjected to Western blot analysis using an antibody against p-TAK1. n=3. Values are presented as the mean±SD. bP<0.05 compared with the normal group; eP<0.05 compared with the LPS treated group.
Discussion
Although the etiology of chronic neurodegenerative diseases such as Alzheimer's disease and Parkinson's disease is unknown, many hypotheses have been proposed for their pathogenesis 25, 26. One such hypothesis is the inflammation hypothesis: the over-activation of microglial cells, as the resident macrophage population in the central nervous system, has been proposed to play a central role in the progression of pathologic inflammation that results in neurodegeneration 10, 11. Compound FLZ is a novel synthetic derivative of squamosamide 1. It has been reported that FLZ has neuroprotective activity against experimental models of Parkinson's disease and promotes learning and memory in animals 5, 6. In the present study, the modulatory effect of FLZ on the LPS-induced production of inflammatory mediators and its underlying mechanism were studied in the macrophage cell line, RAW264.7.
NO is a molecule known to mediate many biological functions, including vasodilation, neurotransmission and host defense 27. The production of NO from L-arginine is catalyzed by different nitric oxide synthases (NOS) 28. NOS-2, commonly called inducible NOS (iNOS), is induced following immunological or inflammatory stimulation in macrophages, astrocytes, microglia and other cell types 29, 30. Frequently, the over-production of NO in macrophages is due to the high expression of iNOS following persistent stimulation. Excessive NO is deleterious and may lead to severe diseases such as septic shock, inflammatory diseases and neurotoxicity 31, 32. The present study demonstrates that pretreatment of RAW264.7 cells with non-toxic concentrations of FLZ (1, 5, or 10 μmol/L) (Table 1) for 30 min significantly inhibited the production of NO and the expression of the iNOS protein and mRNA (Figure 2A, 3).
Sodium nitroprusside hydrolyzes spontaneously, generating NO, which in aqueous solutions at a physiological pH is oxidized quickly to nitrite. In order to demonstrate that FLZ does not directly inhibit NO, SNP was used as a NO donor. FLZ has no effect on nitrite production by SNP (Figure2B), indicating that the effect of FLZ on LPS-induced NO production is the result of the inhibition of iNOS expression.
Prostaglandins are critical factors in inflammation. COX-2 is a key inducible enzyme mediating the biosynthesis of prostaglandins following certain stimuli, including LPS 33. Similar to iNOS, FLZ markedly inhibited the LPS-induced expression of COX-2 protein and mRNA in RAW264.7 cells (Figure 4).
LPS has been shown to initiate the intracellular signaling pathways that lead to the activation of NF-κB 14, 34 and MAPKs 15. NF-κB is an important transcription factor that regulates many genes involved in the inflammatory response, including iNOS and COX-2 35, 36. Following stimulation with LPS, the inhibitory protein IκB is phosphorylated by activated IKKα/β and degraded by the proteasome. This releases NF-κB, which then translocates to the nucleus, where it activates the transcription of multiple genes. The present study demonstrated that FLZ inhibited the increased formation of the NF-κB-DNA complex (Figure 5A) and the nuclear translocation of NF-κB p65 (Figure 5B), as assessed by EMSA and Western blot analyses, respectively. In addition, the phosphorylation and degradation of IκBα were also inhibited by FLZ (Figure 6A, 5C). Phosphorylation of IKKα/β, the upstream kinase of IκB, increased dramatically after stimulation with LPS for 5 min. However, FLZ inhibited this increase in the levels of p-IKKα/β (Figure 6B).
MAPKs are also involved in regulating the production of crucial inflammatory mediators. In response to LPS stimulation, JNK, ERK and p38 MAPK are activated via phosphorylation of both tyrosine and threonine residues, and this phosphorylation leads to the activation of NF-κB 15. The mechanisms by which MAPKs regulate NF-κB-dependent gene expression in LPS-stimulated macrophages are controlled by other sequence-specific transcription factors, such as members of the AP-1 family. In this study, 10 μmol/L FLZ inhibited the LPS-induced increase in the DNA binding activity of AP-1 (Figure 5A) and the phosphorylation of p38 and JNK (Figure 7), but had no effect on ERK phosphorylation (Figure 7). It has been reported that inhibitors of p38 and JNK have anti-inflammatory effects due to inhibition of the expression of inflammatory mediators. Many novel drugs that modulate the activity of the JNK and p38 MAPK pathways have shown promise for treating chronic inflammatory diseases 37, 38. However, compared with JNK and p38 MAPK, there is relatively little information regarding the role of the ERK cascade in inflammatory processes 39. The ERK cascade has been presumed to be the most important pathway for modulating cell proliferation and the upstream molecules that activate ERK should be different than the molecules that activate JNK and p38 MAPK.
LPS is recognized by TLR4 and activates the myeloid differentiation factor 88 (MyD88)-dependent pathway through the MyD88-Interleukin-1-receptor-associated kinase (IRAK)-TNF receptor-associated factor 6 (TRAF6) complex. IRAK-TRAF6, in turn, binds the membrane-bound pre-associated TAK1-TAK1 binding protein (TAB)1-TAB2 complex. The phosphorylation of TAK1 initiates the release of the TAK1-TAB1-TAB2-TRAF6 complex from the membrane and activates TAK1 phosphorylation of downstream targets including IKK, JNK and p38 MAPK 18, 40. Importantly, TAK1 does not seem to be required for the activation of ERK following TLR4 stimulation. This suggests that another molecule is involved in the activation of ERK, and recent reports have demonstrated that Tpl2 is the upstream signaling molecule that mediates the activation of the ERK pathway in response to LPS 41. The inhibition of the phosphorylation of IKKα/β, JNK and p38 MAPK, but not that of ERK, suggests that TAK1 may be one potential upstream target of FLZ. In the present study, various concentrations of FLZ inhibited the LPS-induced phosphorylation of TAK1 (Figure 8). These results suggest that the inhibition of the production of proinflammatory mediators by FLZ may occur through the downregulation of the TAK/IKK and TAK-JNK/p38MAPK pathways.
In one of our recent studies, Zhang et al demonstrated that FLZ reduced the LPS-induced microglial cell production of proinflammatory factors through the inhibition of NADPH oxidase, which is a key microglial superoxide and intracellular ROS-producing enzyme 13. Many studies have demonstrated that increases in intracellular ROS levels can enhance the activation of NF-κB by activating p38 MAPK 41. However, whether ROS are involved in the phosphorylation of TAK1 has not been conclusively demonstrated 40. Furthermore, the in vitro models used in these studies are different and the signaling molecules involved may be different as well. The mechanism by which FLZ inhibits the phosphorylation of TAK1 is worthy of further investigation.
In summary, FLZ inhibited the LPS-induced increase of NO, as well as iNOS and COX-2 expression, at both the RNA and the protein levels in RAW264.7 cells. Additionally, FLZ suppressed the activation of both the TAK1-IKK and the TAK1-JNK/p38MAPK pathways. Finally, FLZ improved learning and memory and the inflammatory response in brain microglia in vivo after intracerebroventricular injection of LPS into mice (data to be reported). Combining our in vitro and in vivo results, we conclude that the protection against neurodegenerative diseases mediated by FLZ may be partially explained by its anti-inflammatory activity.
Author contribution
Geng-tao LIU and Gang LIU designed research; Hong-yan PANG performed research, analyzed data, and wrote the paper.
References
Xie P, Jiao XZ, Liang XT, Feng WH, Wei HL, Liu GT . Synthesis and antioxiactivity of squamosamide cyclic analogs. Zhongguo Yi Xue Ke Xue Yuan Xue Bao 2004; 26: 372–8. Chinese.
Zhang D, Zhang JJ, Liu GT . The novel squamosamide derivative FLZ protects against 6-hydroxydopamine-induced apoptosis through inhibition of related signal transduction in SH-SY5Y cells. Eur J Pharmacol 2007; 561: 1–6.
Zhang D, Zhang JJ, Liu GT . The novel squamosamide derivative (compound FLZ) attenuated 1-methyl-4-phenyl-pyridinium ion (MPP+)-induced apoptosis and alternations of related signal transduction in SH-SY5Y cells. Neuropharmacology 2007; 52: 423–9.
Fang F, Liu GT . Novel squamosamide derivative (compound FLZ) attenuates Abeta(25–35)-induced toxicity in SH-SY5Y cells. Acta Pharmacol Sin 2008; 29: 152–60.
Fang F, Liu GT . Protective effects of compound FLZ on beta-amyloid peptide-(25–35)-induced mouse hippocampal injury and learning and memory impairment. Acta Pharmacol Sin 2006; 27: 651–8.
Feng W, Wei H, Liu GT . Pharmacological study of the novel compound FLZ against experimental Parkinson's models and its active mechanism. Mol Neurobiol 2005; 31: 295–300.
Wyss-Coray T . Inflammation in Alzheimer disease: driving force, bystander or beneficial response? Nat Med 2006; 12: 1005–15.
Gilgun-Sherki Y, Melamed E, Offen D . Anti-inflammatory drugs in the treatment of neurodegenerative diseases: current state. Curr Pharm Des 2006; 12: 3509–19.
McGeer PL, Rogers J, McGeer EG . Inflammation, anti-inflammatory agents and Alzheimer disease: the last 12 years. J Alzheimers Dis 2006; 9: 271–6.
Perry VH, Newman TA, Cunningham C . The impact of systemic infection on the progression of neurodegenerative disease. Nat Rev Neurosci 2003; 4: 103–12.
Block ML, Zecca L, Hong JS . Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci 2007; 8: 57–69.
Heneka MT, O'Banion MK . Inflammatory processes in Alzheimer's disease. J Neuroimmunol 2007; 184: 69–91.
Zhang D, Hu X, Wei SJ, Liu J, Gao H, Qian L, et al. Squamosamide derivative FLZ protects dopaminergic neurons against inflam-mation-mediated neurodegeneration through the inhibition of NADPH oxidase activity. J Neuroinflammation 2008; 5: 21.
Kim YM, Lee BS, Yi KY, Paik SG . Upstream NF-kappaB site is required for the maximal expression of mouse inducible nitric oxide synthase gene in interferon-gamma plus lipopolysaccharide-induced RAW264.7 macrophages. Biochem Biophys Res Commun 1997; 236: 655–60.
Liu Y, Shepherd EG, Nelin LD . MAPK phosphatases — regulating the immune response. Nat Rev Immunol 2007; 7: 202–12.
Sato S, Sanjo H, Takeda K, Ninomiya-Tsuji J, Yamamoto M, Kawai T, et al. Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat Immunol 2005; 6:1087–95.
Cuevas BD, Abell AN, Johnson GL . Role of mitogen-activated protein kinase kinase kinases in signal integration. Oncogene 2007; 26: 3159–71.
Chen ZJ, Bhoj V, Seth RB . Ubiquitin, TAK1 and IKK: is there a connection? Cell Death Differ 2006; 13: 687–92.
Liu Y, Peterson DA, Kimura H, Schubert D . Mechanism of cellular 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction. J Neurochem 1997; 69: 581–93.
Shin HM, Kim BH, Chung EY, Jung SH, Kim YS, Min KR, et al. Suppressive effect of novel aromatic diamine compound on nuclear factor-kappaB-dependent expression of inducible nitric oxide synthase in macrophages. Eur J Pharmacol 2005; 521: 1–8.
Sumanont Y, Murakami Y, Tohda M, Vajragupta O, Matsumoto K, Watanabe H . Evaluation of the nitric oxide radical scavenging activity of manganese complexes of curcumin and its derivative. Biol Pharm Bull 2004; 27: 170–3.
Schreiber E, Matthias P, Muller MM, Schaffner W . Rapid detection of octamer binding proteins with ‘mini-extracts’, prepared from a small number of cells. Nucleic Acids Res 1989; 17: 6419.
Chen C, Chen YH, Lin WW . Involvement of p38 mitogen-activated protein kinase in lipopolysaccharide-induced iNOS and COX-2 expression in J774 macrophages. Immunology 1999; 97: 124–9.
Jeffrey KL, Camps M, Rommel C, Mackay CR . Targeting dual-specificity phosphatases: manipulating MAP kinase signalling and immune responses. Nat Rev Drug Discov 2007; 6: 391–403.
Van Broeck B, Van Broeckhoven C, Kumar-Singh S . Current insights into molecular mechanisms of Alzheimer disease and their implications for therapeutic approaches. Neurodegener Dis 2007; 4: 349–65.
Yuan H, Zheng JC, Liu P, Zhang SF, Xu JY, Bai LM . Pathogenesis of Parkinson's disease: oxidative stress, environmental impact factors and inflammatory processes. Neurosci Bull 2007; 23: 125–30.
Nathan C . Nitric oxide as a secretory product of mammalian cells. FASEB J 1992; 6: 3051–64.
Griffith OW, Stuehr DJ . Nitric oxide synthases: properties and catalytic mechanism. Annu Rev Physiol 1995; 57: 707–36.
Iadecola C, Zhang F, Xu S, Casey R, Ross ME . Inducible nitric oxide synthase gene expression in brain following cerebral ischemia. J Cereb Blood Flow Metab 1995; 15: 378–84.
Lau FC, Bielinski DF, Joseph JA . Inhibitory effects of blueberry extract on the production of inflammatory mediators in lipopolysaccharide-activated BV2 microglia. J Neurosci Res 2007; 85: 1010–7.
Malinski T . Nitric oxide and nitroxidative stress in Alzheimer's disease. J Alzheimers Dis 2007; 11: 207–18.
Zamora R, Vodovotz Y, Billiar TR . Inducible nitric oxide synthase and inflammatory diseases. Mol Med 2000; 6: 347–73.
Minghetti L . Role of COX-2 in inflammatory and degenerative brain diseases. Subcell Biochem 2007; 42: 127–41.
Kim JH, Kim DH, Baek SH, Lee HJ, Kim MR, Kwon HJ, et al. Rengyolone inhibits inducible nitric oxide synthase expression and nitric oxide production by down-regulation of NF-kappaB and p38 MAP kinase activity in LPS-stimulated RAW264.7 cells. Biochemical Pharmacol 2006; 71: 1198–205.
Nomura Y . NF-kappaB activation and IkappaBalpha dynamism involved in iNOS and chemokine induction in astroglial cells. Life Sci 2001; 68: 1695–701.
Guha M, Mackman N . LPS induction of gene expression in human monocytes. Cell Signal 2001; 13: 85–94.
Kaminska B . MAPK signalling pathways as molecular targets for anti-inflammatory therapy-from molecular mechanisms to therapeutic benefits. Biochim Biophys Acta 2005; 1754: 253–62.
Kumar S, Boehm J, Lee JC . p38 MAP kinases: key signalling molecules as therapeutic targets for inflammatory diseases. Nat Rev Drug Discov 2003; 2: 717–26.
Karin M . Inflammation-activated protein kinases as targets for drug development. Proc Am Thorac Soc 2005; 2: 386–90.
Watts C . Location, location, location: identifying the neighborhoods of LPS signaling. Nat Immunol 2008; 9: 343–5.
Su B . Linking stress to immunity? Nat Immunol 2005; 6: 541–2.
Acknowledgements
This work was supported by the Beijing Municipal Natural Science Foundation (grant No 7070001).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Pang, Hy., Liu, G. & Liu, Gt. Compound FLZ inhibits lipopolysaccharide-induced inflammatory effects via down-regulation of the TAK-IKK and TAK-JNK/p38MAPK pathways in RAW264.7 macrophages. Acta Pharmacol Sin 30, 209–218 (2009). https://doi.org/10.1038/aps.2008.29
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2008.29
Keywords
This article is cited by
-
Mungbean seed coat water extract inhibits inflammation in LPS-induced acute liver injury mice and LPS-stimulated RAW 246.7 macrophages via the inhibition of TAK1/IκBα/NF-κB
Journal of Food Science and Technology (2020)
-
Polydatin ameliorates Staphylococcus aureus-induced mastitis in mice via inhibiting TLR2-mediated activation of the p38 MAPK/NF-κB pathway
Acta Pharmacologica Sinica (2017)
-
Atomic force microscopy based investigations of anti-inflammatory effects in lipopolysaccharide-stimulated macrophages
Analytical and Bioanalytical Chemistry (2016)
-
Taurine Attenuates Liver Injury by Downregulating Phosphorylated p38 MAPK of Kupffer Cells in Rats with Severe Acute Pancreatitis
Inflammation (2012)
-
Role of Intrapulmonary Expression of Inducible Nitric Oxide Synthase Gene and Nuclear Factor κB Activation in Severe Pancreatitis-associated Lung Injury
Inflammation (2010)
