
Abstract
Although many thermoelectric materials, such as Bi2Te3, PbTe and CoSb3, possess excellent thermoelectric properties, they often contain toxic and expensive elements. Moreover, most of them are synthesized by processes such as vacuum melting, mechanical alloying or solid-state reactions, which are highly energy and time intensive. All these factors limit commercial applications of the thermoelectric materials. Therefore, it is imperative to develop efficient, inexpensive and non-toxic materials and explore rapid and low-cost synthesis methods. Herein we demonstrated a rapid, facile and low-cost synthesis route that combines thermal explosion (TE) with plasma-activated sintering and used it to prepare environmentally benign CuFeS2+2x. The phase transformation that occurred during the TE and correlations between the microstructure and transport properties were investigated. In a TE process, single-phase CuFeS2 was obtained in a short time and the thermoelectric performance of the bulk samples was better than that of the samples that were synthesized using traditional methods. Furthermore, the effect of phase boundaries on the transport properties was investigated and the underlying physical mechanisms that led to an improvement in the thermoelectric performance were revealed. This work provides several new ideas regarding the TE process and its utilization in the synthesis of thermoelectric materials.
Similar content being viewed by others

Introduction
In the past few decades, increased concerns regarding environmental degradation and rising energy costs have sparked vigorous research activities to identify alternative energy sources and develop novel energy materials. One of the most exciting clean energy conversion technologies is thermoelectricity that can be used to harvest waste industrial heat via the Seebeck effect and convert it into electricity using a purely solid-state means without moving parts1, 2, 3, 4, 5. The efficiency of the conversion process is determined by the dimensionless figure of merit ZT=α2σT /(κe+κL), where α is the Seebeck coefficient, σ is the electrical conductivity, T is the absolute temperature, and κe and κL are the electronic and lattice contributions, respectively, to the thermal conductivity6, 7, 8, 9, 10, 11. Basically, a good thermoelectric material should have both a high Seebeck coefficient and electrical conductivity, and possess as low a thermal conductivity as possible. Currently, there are several families of excellent thermoelectric materials available, such as Bi2Te312, 13, 14, 15, PbTe16, 17, 18, CoSb319, 20, 21, GeTe22, 23, 24, SnSe25, 26 and SnTe27, 28, 29. Although they all possess a high-energy conversion efficiency, most of them contain toxic and expensive elements. Moreover, the synthesis processes that are used consume considerable energy and are time intensive. The above limitations constrain their large-scale industrial applications. Thus, it is important to develop efficient, inexpensive and environmentally friendly thermoelectric materials that can be synthesized by simple, rapid and low-cost routes.
In the past few years, a series of diamond-like structure chalcopyrite compounds including CuInTe230, CuGaTe231, Cu2SnX3 (X=Se, S)32 and Cu3SbX4 (X=Se, S)33 have drawn significant attention due to their unique transport properties. A canonical example is the non-toxic and earth-abundant chalcopyrite compound CuFeS2, which possesses a high Seebeck coefficient (–480 μV K−1 at room temperature)34, 35 and is considered a promising n-type thermoelectric material. Because of the presence of intrinsic sulfur vacancies, CuFeS2 exhibits an n-type semiconducting behavior with a band gap of approximately 0.53 eV36. The chemical states of Cu and Fe in CuFeS2 are monovalent and trivalent, respectively. Owing to the presence of Fe3+, CuFeS2 shows an antiferromagnetic behavior at a Neel temperature of 823 K37, 38. Because of its low electrical conductivity combined with a high lattice thermal conductivity, pure CuFeS2 exhibits a low thermoelectric performance. To improve its thermoelectric performance, most research efforts have been focused on adjusting the carrier concentration by introducing elemental doping,39, 40, 41, 42, 43 controlling the chemical composition34, 44 and introducing sulfur defects36. As the melting points of Cu and Fe are high, the main synthesis approach to obtain CuFeS2 relies on high-temperature melting combined with annealing and this approach requires considerable time and high energy costs. Alternative synthesis methods such as mechanical alloying36, solid-state reactions43, 44 and solution-phase reactions45 also require extensive time and energy. Low thermoelectric performance combined with high fabrication costs have limited commercial applications of this environmentally friendly thermoelectric material.
Recently, researchers at the Wuhan University of Technology46 have developed a combustion synthesis technology that is suitable for the rapid fabrication of many thermoelectric materials, including Bi2Te347, CoSb348, Cu2Se46 and MnSi1.7549, and proposed a new criterion for the applicability of this technology. The latter greatly expands the scope of materials that can be synthesized using the combustion synthesis approach. The benefits of combustion synthesis are dramatically reduced synthesis times and low energy costs. In addtion, the process is easily implemented and readily scalable. Inspired by these beneficial attributes, we attempted to employ the combustion synthesis method for the preparation of CuFeS2 compounds.
According to the ignition modes, the combustion synthesis technique can be divided into a self-propagating high-temperature synthesis, where the ignition is initiated at one end of a cold-pressed pellet of elemental powders, and a thermal explosion (TE) process, where the entire pellet reaches the ignition temperature at the same time. In this work, we used the latter process, TE, to prepare a series of CuFeS2+2x (x=0~0.15) compounds, which were subsequently consolidated and densified via plasma-activated sintering (PAS).
A single-phase CuFeS2 compound was synthesized within 60 s using the TE process and its ZT value was superior to other traditional processing methods for CuFeS2. In addition, the differential scanning calorimetry (DSC) results revealed that the phase transformation of CuFeS2 during the TE synthesis proceeded in two steps: (1) Cu+S→CuS followed by (2) CuS+Fe+S→CuFeS2. However, the phase composition and transport properties of the resulting compound were strongly dependent on the S content, which was considered by expressing the compound as CuFeS2+x. When the actual sulfur content increased, the compound transformed from a pure Cu1.1Fe1.1S2, two-phased region to pure CuFeS2 during the TE and induced changes in the transport properties. As CuFeS2 and Cu1.1Fe1.1S2 are incompatible, the presence of both phases produced a phase boundary which, in turn, had a significant impact on the transport properties. To investigate the effect of the phase boundary on the transport properties, the Bergman-Fel model50, 51 was used to analyze the thermal conductivity, electrical conductivity and Seebeck coefficient, and in the process we demonstrated the critical role of the phase boundary in improving the thermoelectric properties of the composite structure. In addition, we investigated the correlations between the structure, microstructure and transport properties.
Experimental procedures
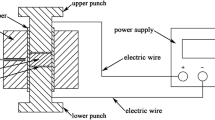
Compounds with the nominal composition of CuFeS2+2x (x=0, 0.05, 0.07, 0.10, 0.12, 0.15 and 0.20) were synthesized by TE combined with the PAS process. High-purity powders of Cu (99.99%, 75 μm), Fe (99.99%, 75 μm) and S (99.99%, 60 μm) were weighed and mixed in stoichiometric proportions to achieve the desired composition (5 g). The mixtures were ground into uniformly fine powders by hand milling, loaded into a steel die and then pressed with a pressure of 10 MPa for 5 min to obtain the pellets. Next, the pellets were sealed in an evacuated quartz tube (diameter of 20 mm) and the tube was subsequently placed into a furnace at a temperature range from 1173 to 1373 K, as shown in Figure 3b. After being held in the furnace at 1173–1373 K for 30–90 s, the quartz tube was taken out and naturally cooled down to room temperature. The obtained products were ground into fine powders and sintered using the PAS apparatus (PAS-III-Ed, Elenix, Japan) under a pressure of 40 MPa at 873 K for 5 min in a vacuum to obtain the densified bulk samples (16 mm diameter × 3.5 mm).
The purity and phase composition of the bulk samples were identified using powder X-ray diffraction analysis (XRD; PANalytical–Empyrean, Almelo, The Netherlands: X’Pert PRO, Cu Kα). The morphology of the bulk samples was studied using a field-emission scanning electron microscopy (Hitachi SU8020, Tokyo, Japan) and the chemical compositions were obtained with an energy-dispersive spectrometer (JXA-8230/INCAX-ACT, Tokyo, Japan). The heat flow during the TE process was measured by power compensation DSC (DSC Q20; TA Instrument, Delaware, PA, USA). The apparatus was calibrated before testing. The electrical conductivity and Seebeck coefficient were measured using a ZEM-3 apparatus (Ulvac Riko, Inc., Kanagawa, Japan) under a helium atmosphere from 300 to 625 K. The thermal conductivity was calculated from the relationship κ=DCpρ, where D, Cp and ρ are the thermal diffusivity, the heat capacity and the density of bulk samples, respectively. The thermal diffusivity was measured using a laser-flash system (LFA 457; Netzsch, Selb, Germany) in an argon atmosphere. The heat capacity and the heat flow were determined by power compensation DSC (DSC Q20; TA Instrument) and the sample density was specified by the Archimedes’ method. The room-temperature Hall coefficient (RH) was measured using a physical property measurement system (PPMS-9: Quantum Design, San Diego, CA, USA). The carrier concentration (n) and carrier mobility (μH) were calculated according to n=1/eRH and μH=σ/ne, respectively. We estimated the overall accuracy of the measured ZT values to be approximately ±10%.
Results and discussion
The phase transformation mechanism of CuFeS2 during TE synthesis
The TE synthesis reaction is a type of combustion synthesis where the whole sample reaches the ignition temperature at the same time and the reaction simultaneously begins within the entire sample. To determine whether CuFeS2 can be synthesized by TE and how the formation of CuFeS2 proceeded during the TE synthesis, DSC was employed to track the heat flow during the process. High-purity powders of Cu (99.99%), Fe (99.99%) and S (99.99%) were weighed, mixed in stoichiometric proportions and ground into uniformly fine powders by hand milling and then cold-pressed to obtain the raw pellets. The pellets were loaded into the DSC apparatus, heated to high temperatures of 423, 758 and 823 K under a nitrogen atmosphere at a heating rate of 30 K min−1. Subsequently, the phase compositions of the products were identified using powder XRD analysis. The results are shown in Figures 1 and 2. First, we tested whether CuFeS2 can be synthesized by TE by heating the pellet in the DSC apparatus to 823 K. The heat flow curve and XRD pattern of the resulting product are shown in Figure 1. In this case, two intense exothermic signatures at 394 and 758 K, and two endothermic markers at 378 and 781 K were observed. The XRD pattern of the product revealed that, after heating to 823 K, the pellet reacted and its phase composition was CuFeS2 with FeS2 and Cu5FeS4 secondary phases. This result confirms that CuFeS2 can be synthesized by TE and the DSC scan is an effective method to trace the phase transformation process during TE.

(a) DSC curve of Cu-Fe-S=1:1:2 mixed powders that were pellet heated to 823 K; (b) XRD pattern of the product after heating to 823 K using DSC.

(a) Step 1: DSC curve of Cu-Fe-S=1:1:2 mixed powders that were pellet heated to 423 K; (b) Step 2 DSC curve of Cu-Fe-S=1:1:2 mixed powders that were pellet heated to 758 K; (c) XRD pattern of the product after Step 1 and (d) XRD pattern of the product after Step 2.
To investigate the exothermic phenomena at 394 and 758 K, two experiments were performed as follows.
In Step 1, the DSC apparatus was used to heat a new raw pellet to 423 K at a rate of 30 K min−1 and then the pellet was quickly cooled to room temperature. The results of this procedure are shown in Figures 2a and c. As the pellet’s temperature reached 379 K, an endothermic peak was observed in the DSC curve, which corresponds to the melting point of sulfur. When the pellet’s temperature reached 392 K, an intense exothermic peak was detected, which signals that an intense exothermic reaction occurred at 392 K. The corresponding XRD pattern indicates that the pellet consists of CuS, Fe and a small amount of S. Combining the DSC data with the XRD pattern, we concluded that when a raw pellet is heated to 392 K, Cu reacts with S to form CuS, whereas unreacted Fe and elemental S remained. We interpreted this as the first step in the formation of CuFeS2 during the TE process.
In Step 2, another new raw pellet was heated in the DSC apparatus from room temperature to 758 K with a heating rate of 30 K min−1. The corresponding DSC curve is shown in Figure 2b. Apart from the endothermic peak (the melting of sulfur at temperature T1=379 K) and the exothermic peak (the formation of CuS at temperature T2=392 K) discussed above, another intense exothermic peak was observed on the DSC curve at 758 K. Figure 2d shows the powder XRD pattern of the pellet after heating to 758 K. Evidently, the pellet had fully reacted and turned into CuFeS2. Thus, we conclude that after heating to 758 K, the CuS compound that formed at 393 K reacted with S and Fe to produce the CuFeS2 compound. The formation of CuFeS2 in the TE process is therefore a two-step transformation. Finally, according to the DSC results and literature data52, the endothermic signal observed at 781 K in Figure 1 represents an oxidation–reduction reaction between CuFeS2 and excess S, as shown in equation (4). The sequence of reactions that occurred during the TE synthesis of CuFeS2 is summarized by the following equations.




As reactions (2) and (3) are intense exothermic reactions, they enable the synthesis of CuFeS2 via a TE.
TE parameters for CuFeS2
According to the DSC data, the formation of the CuFeS2 compound via a TE requires the heating of a pellet to at least 758 K, but not much higher, because the temperature of the oxidation-reduction reaction occurs at temperatures above 781 K. Therefore, the synthesis of CuFeS2 is sensitive to the TE parameters. The TE temperature should be high enough to ensure that the raw pellet is ignited and high heating rate is maintained; however, the duration should be short so that the pellet is not overheated. To determine the appropriate TE parameters for CuFeS2, a series of compounds were synthesized at various temperatures (1173, 1273 and 1373 K) and holding times (20, 30, 40, 60 and 90 s) via the TE method using a furnace. The synthesis method of TE, as discussed in the experiment section and the respective XRD patterns of the products are shown in Figure 3a.

(a) XRD patterns of the CuFeS2 samples that were synthesized using TE at various temperatures and different holding times and (b) schematic description of the TE process.
When the TE temperature was 1173 K and the holding time was 20 s, an XRD scan indicated the presence of only CuS. The reason is because of a short holding time during which the pellet was unable to reach temperatures above 758 K, even though the temperature of the furnace was 1173 K. As the holding time was increased to 40 and 60 s, the presence of CuFeS2 was detected on the XRD scan together with a few secondary phases (see Figure 3a). Thus, the temperature of 1173 K is too low to achieve a sufficiently high heating rate and what was to be the TE process turned into a solid-state reaction, which requires a long annealing time to form a homogeneous product on an atomic scale.
When the furnace temperature was increased to 1273 K and the holding time was either 30 or 60 s, a single-phase CuFeS2 compound was obtained (Figure 3a). When the holding time was increased to 90 s, a second phase of Cu5FeS4 appeared in the XRD pattern. This phase was interpreted as an overheated pellet that began to decompose because of an extensive holding time. In addition, with an increase in the furnace temperature to 1373 K and the use of 20 and 60 s holding times, the content of Cu5FeS4 and S increased in the pellet, which means that the TE temperature was too high and the pellet was severely decomposing. After this series of experiments, we concluded that a temperature of 1273 K and a holding time of 60 s promoted the TE synthesis of CuFeS2 (without decomposing) in a typical 5 g pellet.
Phase composition and thermoelectric property
Owing to low melting and boiling points, S readily volatilizes during TE, which can lead to deviations from the nominal stoichiometric compositions. Such variations in S content can influence the transport properties of the materials. In this section, we attempt to adjust the content of sulfur in CuFeS2 and correlate the thermoelectric properties with the phase composition.
Phase composition and microstructure characterization
A series of CuFeS2+2x (x=0, 0.05, 0.07, 0.10, 0.12, 0.15 and 0.20) compounds were synthesized using TE combined with the PAS process under the optimal conditions that were described in the previous section. Powder XRD patterns of the respective final products are shown in Figure 4a. When the excess sulfur exceeded x⩽0.15, then pure CuFeS2 was obtained. When x⩾0.20, secondary phases of Cu5FeS4 and FeS2 were present in the final products. This result indicates that when x⩾0.20, there is excessive sulfur in the starting compositions, which leads either to an oxidation–reduction reaction of CuFeS2 or the preferential formation of the Cu5FeS4 and FeS2 phases. Figure 4b shows powder XRD patterns of samples prepared using TE followed by PAS sintering. There are no substantive changes compared with the patterns in Figure 4a, which depicts XRD patterns of the same compounds before PAS sintering. Again, when x=0–0.15, pure CuFeS2 was obtained, whereas for the x=0.20 sample, the intensities of the Cu5FeS4 and FeS2 peaks increased. These data suggest that excessive S can exist in the TE product and lead to an oxidation–reduction reaction between CuFeS2 and excess S during PAS sintering. Moreover, the loss of sulfur during the reaction should not be compensated for with greater than x=0.15 nominal excess S in the starting material. Figure 4c shows the main XRD peak (2θ=29°) as a function of composition x. As the content of sulfur is varied, two phases, Cu1.1Fe1.1S2 and CuFeS2, are detected in the compounds. As sulfur easily evaporates during TE, the nominal x=0 sample was a sulfur-deficient material. These data in Figure 4c suggest that its composition was Cu1.1Fe1.1S2. As the content of sulfur increased (x=0.05-0.12), the actual deficiency of S was diminished and lead to a gradual phase transformation from Cu1.1Fe1.1S2 to CuFeS2, which was represented by the appearance of a double-peak structure. This structure suggests that the material phase separated into Cu1.1Fe1.1S2 and CuFeS2 phases rather than forming a solid solution. Finally, when x=0.15, a single pure CuFeS2 phase was obtained.

Powder XRD patterns of CuFeS2+2x (x=0–0.20) prepared using (a) TE and (b) TE combined with PAS sintering, and (c) the main XRD peak as a function of composition x.
Microstructures of freshly fractured surfaces of all sintered samples were examined using field-emission scanning electron microscopy. As the morphology of all samples was similar, the x=0 and x=0.15 compounds were chosen as examples and shown in Figure 5. The PAS-sintered compounds were fully dense with relative densities above 98%. The fracture mechanism was a trans-crystalline rupture. No obvious grains or grain boundaries were observed; however, there were numerous 100–300 nm nanoholes, which were uniformly distributed on the smooth surfaces of the matrix grains. Figure 6 shows back-scattered electron images of a polished surface of CuFeS2+2x. No distinct impurity phases were detected when x=0. When x=0.05, numerous stripe-type secondary-phase regions emerged that were uniformly distributed in the matrix. As described above, the compound was in a two-phase composition region. When the sulfur content x increased, the volume of the secondary phase increased, whereas that of the matrix decreased. When x=0.15, the compound was a pure-phase (CuFeS2) and no trace of the impurity phase was detected. The actual chemical composition of all samples that was determined using energy-dispersive spectrometer measurements is presented in Table 1. From the energy-dispersive spectrometer data and XRD results, as the S content increased, the phase composition evolved from Cu1.1Fe1.1S2 to CuFeS2 and the phase boundaries between the two compounds were present. DSC heat-flow curves that were obtained on the PAS-sintered samples are shown in Figure 7. A reversible phase transition near 490 K was observed for the x=0 compound (Cu1.1Fe1.1S2). As the sulfur content x increased, the intensity and peak area, which characterized this phase transition, decreased. When x=0.15, the peak completely disappeared, which indicates that the phase transition from Cu1.1Fe1.1S2 to CuFeS2 was complete. The DSC approach is more sensitive than the XRD or field-emission scanning electron microscopy methods and using the DSC peak area to determine the phase content in a mixture is a very reliable and mature technique. The relative amount of Cu1.1Fe1.1S2 and CuFeS2 phases in the compounds can be assessed from the area of the characteristic peak and the results are presented in Table 1.

Field-emission scanning electron microscopy (FESEM) images of the free fracture surface of PAS-sintered CuFeS2+x (x=0 and 0.15).

BSE images of the polished surfaces for PAS-sintered CuFeS2+x (x=0, 0.05, 0.07, 0.10, 0.12 and 0.15).

DSC curves of PAS-sintered CuFeS2+x (x=0, 0.05, 0.07, 0.10, 0.12 and 0.15).
According to the XRD, energy-dispersive spectrometer and DSC results that are presented in this section, the following conclusions can be drawn. There are two phases in the CuFeS2+2x compound, which was prepared using TE. The nominal stoichiometric CuFeS2 compound (x=0) is a sulfur-poor structure of composition Cu1.1Fe1.1S2 due to the loss of sulfur during the thermal-explosion reaction. As the S content increased, a two-phase region, which consists of a mixture of Cu1.1Fe1.1S2 and CuFeS2 phases, was entered. The phase transformation from Cu1.1Fe1.1S2 to CuFeS2 was completed when the excess sulfur attained x=0.15. Attempts to impose a greater excess of sulfur into the structure led to the decomposition of CuFeS2 into FeS2 and Cu5FeS4.
Electrical transport properties
The room temperature transport properties of all compounds are summarized in Table 2. As previously stated, the nominally stoichiometric CuFeS2 (x=0) was S deficient because of S loss during the TE synthesis. To compensate for this loss by increasing the S content x, the deficiency diminished and this was reflected in the decreasing carrier concentration and electrical conductivity, whereas the Seebeck coefficient and carrier mobility increased. The temperature dependence of the electrical and thermal transport properties is discussed below.
The temperature dependence of the electrical conductivity of all samples is shown in Figure 8a. Because of the decreasing carrier concentration, electrical conductivity at room temperature markedly decreased from 1.9 × 104 Sm−1 in Cu1.1Fe1.1S2 to 2.9 × 103 Sm−1 in CuFeS2. For pure CuFeS2 (x=0.15), the electrical conductivity exhibited a semiconductor behavior, whereas the samples with a S content of x=0-0.12 (a mixture of Cu1.1Fe1.1S2 and CuFeS2 phases), the electrical conductivity initially (300–475 K) increased with increasing temperature and then reversed at ~475 K due to the phase transition of Cu1.1Fe1.1S2. Figure 8b shows that all samples exhibited negative Seebeck coefficients over the entire temperature range, which documents the n-type character of the transport with electrons as the dominant charge carrier. With an increase in S content that was accompanied by a decreasing electron density, the absolute value of the Seebeck coefficient increased. In pure CuFeS2 (x=0.15), the absolute value of the Seebeck coefficient monotonically decreased with an increase in temperature, whereas in samples with x=0–0.12, the absolute value of the Seebeck coefficient increased to ~450 K and then decreased. Combining the good electrical conductivity of Cu1.1Fe1.1S2 with the high Seebeck coefficient of CuFeS2, the power factor α2σ of the mixtures of the phases (x=0.05-0.12) was higher than that of each of the pure Cu1.1Fe1.1S2 (x=0) and CuFeS2 (x=0.15) phases. The highest power factor in the covered temperature range was obtained when x=0.12, as shown in Figure 8c.

The temperature dependence of the (a) electrical conductivity, (b) Seebeck coefficients and (c) power factor for CuFeS2+x (x=0, 0.05, 0.07, 0.10, 0.12 and 0.15).
Thermal conductivity
Figure 9a shows the temperature dependence of the total thermal conductivity for all samples. As the temperature increased, the total thermal conductivity significantly decreased because of the enhanced contribution of the Umklapp processes. In addition, due to the phase transition of Cu1.1Fe1.1S2 at 490 K, all transport properties behave an abnormal change in the phase transition region, as shown in Supplementary Figure S1 in the Supplementary Information. As the S excess x increased, the total thermal conductivity systematically and dramatically increased due to the high thermal conductivity of the progressively abundant CuFeS2 phase. Figure 9b shows the temperature dependence of the lattice thermal conductivity. As contributions from the bipolar thermal conductivity are small within the investigated temperature range, we can estimate the lattice thermal conductivity by simply subtracting the electronic thermal conductivity from the measured total thermal conductivity,8

Temperature dependence of the (a) total thermal conductivity and (b) lattice thermal conductivity for CuFeS2+x (x=0, 0.05, 0.07, 0.10, 0.12 and 0.15).

where κL, κ and κe are the lattice thermal conductivity, total thermal conductivity and electronic thermal conductivity, respectively. The latter can be estimated using the Wiedemann–Franz relation: κe=LσT, where σ is the electrical conductivity and L the Lorenz number, which can be extracted by the fitting of the respective Seebeck coefficient values. Although the obtained samples were a mixture when x=0.05–0.12, there is not an effective model for composites to calculate the Lorenz number; therefore, equations (6) and (7) were used to approximately determine the its value. Assuming a single parabolic band model and acoustic phonon scattering as the main carrier-scattering mechanism, the Lorenz number L is calculated by53, 54

Here, the reduced Fermi energy η can be obtained from the value of the Seebeck coefficient34


Here, kB, e and α are the Boltzmann constant, the charge of an electron and the Seebeck coefficient, respectively, and r is the scattering factor. Here, r=−1/239. Thus, the obtained lattice thermal conductivity is shown in Figure 9b. Because of the small magnitude of the electrical conductivity, the carrier contribution to the total thermal conductivity was small. The room temperature lattice thermal conductivity increased with increasing S content from 1.7 Wm−1 K−1 for pristine Cu1.1Fe1.1S2 (x=0) to 6.4 Wm−1 K−1 for CuFeS2 (x=0.15). This change was primarily because the crystal structure of the Cu1.1Fe1.1S2 compound (Cubic, I-43m, a=b=c=10.603 Å) is more complex than that of the CuFeS2 compound (Tetragonal, I-42d, a=b=5.2854 Å), which possesses a diamond-like crystal structure (the crystal structure iss hown in Supplementary Figure S2 in the Supplementary Information). The complex crystal structure of the Cu1.1Fe1.1S2 compound increased the bond length and introduced diverse chemical bonding in the structure and the diverse chemical bonding led to a low thermal conductivity. In addition, in the structure of Cu1.1Fe1.1S2, there were several voids (Site: 6b, 24 g) and some of the voids were partially filled by Cu and Fe. It is possible that those partially filled atoms rattled in the void and caused intense phonon scattering. All these factors contributed to the low thermal conductivity and led to the different lattice thermal conductivities of the two compounds. As the S content increased from x=0 to x=0.15, the phase composition varied from pure Cu1.1Fe1.1S2 to a two-phase mixture and to pure CuFeS2, which resulted in a progressively increasing lattice thermal conductivity.
The effect of the phase boundary on transport properties
To study the effect of the phase boundary between the Cu1.1Fe1.1S2 and CuFeS2 phases on the transport properties, an effective medium approximation model proposed by Bergman and Fel50 was applied to analyze the electrical and thermal transport properties. Furthermore, the theoretical and experimental results were compared with gain insight into the influence of temperature on the transport properties of the two-phase structures. Based on the Bergman-Fel model50, 51, 55 for a random dispersion of component B embedded in a matrix A, the effective electrical conductivity σE, the effective thermal conductivity κE and the effective Seebeck coefficient αE were obtained by fitting the electrical conductivities σA and σB, thermal conductivities κA and κB and Seebeck coefficients αAand αB of the two components A and B using the following expressions.



Here, Φ is the volume fraction of component B and the other parameters are given by equations (12)–(14), as follows.



From the transport measurements and DSC analysis, the thermoelectric transport properties and phase contents of components A (Cu1.1Fe1.1S2) and B (CuFeS2) were obtained, which allow predictions regarding the relationship between the effective transport property and temperature that are based on the above calculations. The results are displayed in Figure 10. The overall trend and magnitude of the calculated data were comparable to the experimental results, which confirms that the transport properties of the samples are consistent with their composite character. Moreover, the results verify that the Cu1.1Fe1.1S2 and CuFeS2 component compounds were phase separated and independent.

The calculated results based on the Bergman-Fel model: The temperature dependence of the (a) electrical conductivity, (b) Seebeck coefficients and (c) thermal conductivity for CuFeS2+x (x=0, 0.05, 0.07, 0.10, 0.12 and 0.15).
Although the Bergman–Fel model can estimate the transport properties of a composite medium, the model does not take into account the phase boundary scattering and electron redistribution between different components that may arise and lead to deviations between the calculated and experimental values. Here, we define a deviation parameter Δx (x=σ, α, κ) to calculate the discrepancies between the theory and experiments, and obtain an estimate of the influence of phase boundaries on the transport properties. The deviation parameter Δx (x=σ, α, κ) is defined as:

The respective deviation parameters for the electrical conductivity, Seebeck coefficient and thermal conductivity are shown in Figure 11a. A positive number means that the experimental value overestimates the theory, whereas a negative number implies that the experimental data are smaller than the theoretical estimates. First, we observed that the experimental data for the thermal conductivity of all samples were lower than the theoretical estimates. However, as the temperature increased, the deviation between the experiment and theory significantly decreased. As already noted, the Bergman–Fel model does not take into account phonon scattering at the phase boundaries in the thermal conductivity calculations. At room temperature, the mean phonon energy was low and these low-frequency phonons provide a significant contribution to the heat transport. However, the presence of phase boundaries can efficiently scatter these low-frequency phonons, as illustrated in Figure 11b, and this can lead to the additional suppression of the thermal conductivity, which causes a large (negative) deviation with respect to the calculated values. As the temperature increased, the mean phonon energy increased and these higher frequency phonons were less effectively scattered by the phase boundaries, which decreased the deviation between the experimental results and theoretical estimates. This result is consistent with a general understanding that phase-boundary scattering more effectively reduces the thermal conductivity at low rather than high temperatures.

(a) Deviations between the experimental and calculated data based on the Bergman–Fel model; (b) schematic description of phonon scattering at the phase boundaries at low and high temperatures and (c) schematic description of the electron redistribution at the phase boundary.
Although electrons are also expected to be scattered by the phase boundaries, which would lead to a decreased electrical conductivity, the actual experimental values of the electrical conductivity and Seebeck coefficient are larger than the respective theoretical predictions (positive Δσ and Δα). The reason may be because the Bergman–Fel model does not take into account the carrier redistribution in calculating the composite transport properties and regards them as a mixture of phases. From the Hall effect measurements, we know that the carrier concentration of Cu1.1Fe1.1S2 (n=9.8 × 1020 cm−3) was much higher than that of CuFeS2 (n=1.8 × 1019 cm−3), which indicates that the Fermi levels of CuFeS2 and Cu1.1Fe1.1S2 are vastly different. Consequently, upon the contact of the two phases, a significant electron transfer is expected from Cu1.1Fe1.1S2 to CuFeS2, which results in a major redistribution of charge carriers in the proximity of the interface, as schematically depicted in Figure 11c. In this case, the Fermi level imbalance prompted electron diffusion from the Cu1.1Fe1.1S2 phase to the CuFeS2 phase with the latter possessing a higher carrier concentration than the concentration of its ionized donor impurity centers would imply. Consequently, the higher carrier concentration with a lower density of ionized donors (in comparison to a uniformly doped structure) demonstrated effects that are similar to that of modulation doping56, 57, 58 and the CuFeS2 matrix phase should have a higher carrier mobility. Thus, the overall electrical conductivity of the composite should be enhanced.
With an increasing S content, the difference between the actual and theoretical electrical conductivity values increased, as shown in Figure 11a. This effect is due to the increasing S content that promotes the formation of the CuFeS2 phase, which effectively enhances the modulation-like doping because the carrier mobility of CuFeS2 (μ=10 cm2 V−1 s−1) is much larger than that of Cu1.1Fe1.1S2 (μ=1.2 cm2 V−1 s−1). However, the actual electrical conductivity as a function of temperature became closer to the theoretical value at elevated temperatures. This is likely to be due to the increasing dominance of acoustic phonon scattering at higher temperatures, which will particularly affect the mobility of the higher mobility phase (CuFeS2), weaken the modulation-like doping and reduce the deviation parameter Δσ. The carrier concentration redistribution may also alter the band structure and band alignment of the two phases and give rise to deviations in the Seebeck coefficient between the experiments and theory.
Finally, we considered the effect of the phase boundaries on the thermoelectric figure of merit. Figure 12 depicts the temperature dependence of the figure of merit ZT of our TE-synthesized CuFeS2+2x compounds and, for comparison, we also included the literature data for samples that were fabricated by different synthesis routes. By combining the good electrical conductivity of the Cu1.1Fe1.1S2 phase with the high Seebeck coefficient of the CuFeS2 phase, the x=0.05 compound possesses a high-power factor coupled with a low thermal conductivity, which results in an improved ZT value of 0.23 that was achieved at 625 K. This represents an ~130% enhancement over the value for the nominally pristine CuFeS2 sample (x=0). Compared with the CuFeS2 compound that was synthesized by other methods, notably vacuum melting34, mechanical alloying36 or a solid-state reaction43, the thermoelectric performance of the compound that was synthesized by TE followed by PAS sintering was vastly superior. Equally important, the TE synthesis consumed substantially less energy and required a short period to implement.

Temperature dependence of the figure of merit ZT for CuFeS2+x (x=0, 0.05, 0.07, 0.10, 0.12 and 0.15) and the samples synthesized by other methods. MA denotes mechanical alloying and solid-state reaction represents the compounds that were synthesized by solid-state reactions.
Conclusions
CuFeS2 compounds have been successfully synthesized using TE combined with the PAS-sintering process. The formation of the CuFeS2 phase during the TE synthesis proceeded in two steps: (1) Cu+S→CuS (at ~392 K) followed by (2) CuS+Fe+S→CuFeS2 (at ~758 K). As sulfur readily volatilizes during the TE process, a series of CuFeS2+2x (x=0~0.15) compounds were synthesized to compensate for the loss of sulfur and investigate the correlations between the structure, microstructure and transport properties. The results revealed that two phases form during the TE synthesis, which depend on the exact S content, reaction temperature and holding time. When the final reaction product attained an elemental composition ratio of Cu:Fe:S=1:1:2 (that required an excess of sulfur in the starting mixture of the elemental powders of x=0.15), pure-phase CuFeS2 was obtained. When the excess sulfur in the starting mixture was reduced, 0.05⩽ × ⩽0.12, there was not enough sulfur to compensate for its evaporation during the TE and hence the resulting product contained a mixture of CuFeS2 and Cu1.1Fe1.1S2 phases with a progressively higher fraction of the latter. Eventually, with no excess sulfur in the starting elemental powder mixture (x=0), only the sulfur-deficient Cu1.1Fe1.1S2 phase was formed. The two CuFeS2 and Cu1.1Fe1.1S2 phases were incompatible, which produced phase boundaries between the two phase components. The presence of phase boundaries affected the transport properties by scattering low-frequency phonons and reducing the thermal conductivity of the composite structure. Moreover, the large differences in the carrier concentration (electrons) in CuFeS2 and Cu1.1Fe1.1S2 facilitated a redistribution of electrons in the composite and led to an enhancement in the electronic transport properties. Combining the good electrical conductivity of the Cu1.1Fe1.1S2 phase with the high Seebeck coefficient of the CuFeS2 phase, the compound with x=0.05 possessed the highest ZT value of 0.23, which was achieved at 625 K. This value represents an ~130% enhancement over the value for the pristine CuFeS2 compound (x=0.15) and is superior to the values for the CuFeS2 compounds that were prepared using other synthesis routes. Furthermore, TE is a rapid synthesis process that consumes significantly less energy than alternative fabrication processes and is easily implemented. By demonstrating several attributes of a TE synthesis reaction, we hope that it may be adopted as an efficient, low-cost and rapid processing technique for other environmentally friendly and inexpensive thermoelectric materials.
References
Sootsman, J. R., Chung, D. Y. & Kanatzidis, M. G. New and old concepts in thermoelectric materials. Angew. Chem. Int. Ed. 48, 8616–8639 (2009).
Bell, L. E. Cooling, heating, generating power, and recovering waste heat with thermoelectric systems. Science 321, 1457–1461 (2008).
Dresselhaus, M. S., Chen, G., Tang, M. Y., Yang, R. G., Lee, H., Wang, D. Z., Ren, Z. F., Fleurial, J. P. & Gogna, P. New directions for low-dimensional thermoelectric materials. Adv. Mater. 19, 1043–1053 (2007).
Fu, C., Bai, S., Liu, Y., Tang, Y., Chen, L., Zhao, X. & Zhu, T. Realizing high figure of merit in heavy-band p-type half-Heusler thermoelectric materials. Nat. Commun. 6, 8144 (2015).
Xie, H., Wang, H., Pei, Y., Fu, C., Liu, X., Snyder, G. J., Zhao, X. & Zhu, T. Beneficial contribution of alloy disorder to electron and phonon transport in half-Heusler thermoelectric materials. Adv. Funct. Mater. 23, 5123–5130 (2013).
Lin, S., Li, W., Chen, Z., Shen, J., Ge, B. & Pei, Y. Tellurium as a high-performance elemental thermoelectric. Nat. Commun. 7, 10287 (2016).
He, J., Kanatzidis, M. G. & Dravid, V. P. High performance bulk thermoelectrics via a panoscopic approach. Mater. Today 16, 166–176 (2013).
Snyder, G. J. & Toberer, E. S. Complex thermoelectric materials. Nat. Mater. 7, 105–114 (2008).
Tritt, T. M. & Subramanian, M. A. Thermoelectric materials, phenomena, and applications: a bird’s eye view. MRS Bull. 31, 188–198 (2006).
Zhu, T., Gao, H., Chen, Y. & Zhao, X. Ioffe–Regel limit and lattice thermal conductivity reduction of high performance (AgSbTe2 15(GeTe)85 thermoelectric materials. J. Mater. Chem. A 2, 3251–3256 (2014).
Su, X., Wei, P., Li, H., Liu, W., Yan, Y., Li, P., Su, C., Xie, C., Zhao, W., Zhai, P., Zhang, Q., Tang, X. & Uher, C. Multi-scale microstructural thermoelectric materials: transport behavior, non-equilibrium preparation, and applications. Adv. Mater. 1602013 (2017).
Zheng, Y., Zhang, Q., Su, X., Xie, H., Shu, S., Chen, T., Tan, G., Yan, Y., Tang, X., Uher, C. & Snyder, G. J. Mechanically robust BiSbTe alloys with superior thermoelectric performance: a case study of stable hierarchical nanostructured thermoelectric materials. Adv. Energy Mater. 5, 1401391 (2015).
Xie, W., He, J., Kang, H. J., Tang, X., Zhu, S., Laver, M., Wang, S., Copley, J. R., Brown, C. M., Zhang, Q. & Tritt, T. M. Identifying the specific nanostructures responsible for the high thermoelectric performance of (Bi,Sb)2Te3 nanocomposites. Nano Lett. 10, 3283–3289 (2010).
Xu, Z., Wu, H., Zhu, T., Fu, C., Liu, X., Hu, L., He, J., He, J. & Zhao, X. Attaining high mid-temperature performance in (Bi,Sb)2Te3 thermoelectric materials via synergistic optimization. NPG Asia Mater. 8, e302 (2016).
Hu, L.-P., Zhu, T.-J., Wang, Y.-G., Xie, H.-H., Xu, Z.-J. & Zhao, X.-B. Shifting up the optimum figure of merit of p-type bismuth telluride-based thermoelectric materials for power generation by suppressing intrinsic conduction. NPG Asia Mater. 6, e88 (2014).
Biswas, K., He, J., Blum, I. D., Wu, C. I., Hogan, T. P., Seidman, D. N., Dravid, V. P. & Kanatzidis, M. G. High-performance bulk thermoelectrics with all-scale hierarchical architectures. Nature 489, 414–418 (2012).
Pei, Y., Shi, X., LaLonde, A., Wang, H., Chen, L. & Snyder, G. J. Convergence of electronic bands for high performance bulk thermoelectrics. Nature 473, 66–69 (2011).
Pei, Y., Wang, H., Gibbs, Z. M., LaLonde, A. D. & Snyder, G. J. Thermopower enhancement in Pb1−xMnxTe alloys and its effect on thermoelectric efficiency. NPG Asia Mater. 4, e28 (2012).
Su, X., Li, H., Yan, Y., Chi, H., Tang, X., Zhang, Q. & Uher, C. The role of Ga in Ba0.30GaxCo4Sb12+x filled skutterudites. J. Mater. Chem. 22, 15628 (2012).
Su, X., Li, H., Wang, G., Chi, H., Zhou, X., Tang, X., Zhang, Q. & Uher, C. Structure and transport properties of double-doped CoSb2.75Ge0.25–xTex (x= 0.125–0.20) with in situ nanostructure. Chem. Mater. 23, 2948–2955 (2011).
Liang, T., Su, X., Yan, Y., Zheng, G., She, X., You, Y., Uher, C., Kanatzidis, M. G. & Tang, X. Panoscopic approach for high-performance Te-doped skutterudite. NPG Asia Mater. 9, e352 (2017).
Perumal, S., Roychowdhury, S., Negi, D. S., Datta, R. & Biswas, K. High thermoelectric performance and enhanced mechanical stability of p-type Ge1–xSbxTe. Chem. Mater. 27, 7171–7178 (2015).
Wu, D., Zhao, L. D., Hao, S., Jiang, Q., Zheng, F., Doak, J. W., Wu, H., Chi, H., Gelbstein, Y., Uher, C., Wolverton, C., Kanatzidis, M. & He, J. Origin of the high performance in GeTe-based thermoelectric materials upon Bi2Te3 doping. J. Am. Chem. Soc. 136, 11412–11419 (2014).
Wu, L., Li, X., Wang, S., Zhang, T., Yang, J., Zhang, W., Chen, L. & Yang, J. Resonant level-induced high thermoelectric response in indium-doped GeTe. NPG Asia Mater. 9, e343 (2017).
Zhao, L.-D., Tan, G., Hao, S., He, J., Pei, Y., Chi, H., Wang, H., Gong, S., Xu, H., Dravid, V. P., Uher, C., Snyder, G. J., Wolverton, C. & Kanatzidis, M. G. Ultrahigh power factor and thermoelectric performance in hole-doped single-crystal SnSe. Science 351, 141–144 (2015).
Zhao, L. D., Lo, S. H., Zhang, Y., Sun, H., Tan, G., Uher, C., Wolverton, C., Dravid, V. P. & Kanatzidis, M. G. Ultralow thermal conductivity and high thermoelectric figure of merit in SnSe crystals. Nature 508, 373–377 (2014).
Banik, A., Vishal, B., Perumal, S., Datta, R. & Biswas, K. The origin of low thermal conductivity in Sn1−xSbxTe: phonon scattering via layered intergrowth nanostructures. Energy Environ. Sci. 9, 2011–2019 (2016).
Tan, G., Shi, F., Doak, J. W., Sun, H., Zhao, L.-D., Wang, P., Uher, C., Wolverton, C., Dravid, V. P. & Kanatzidis, M. G. Extraordinary role of Hg in enhancing the thermoelectric performance of p-type SnTe. Energy Environ. Sci. 8, 267–277 (2015).
Zhang, Q., Liao, B., Lan, Y., Lukas, K., Liu, W., Esfarjani, K., Opeil, C., Broido, D., Chen, G. & Ren, Z. High thermoelectric performance by resonant dopant indium in nanostructured SnTe. Proc. Natl Acad. Sci. USA 110, 13261–13266 (2013).
Liu, R., Xi, L., Liu, H., Shi, X., Zhang, W. & Chen, L. Ternary compound CuInTe2: a promising thermoelectric material with diamond-like structure. Chem. Commun. 48, 3818–3820 (2012).
Plirdpring, T., Kurosaki, K., Kosuga, A., Day, T., Firdosy, S., Ravi, V., Snyder, G. J., Harnwunggmoung, A., Sugahara, T., Ohishi, Y., Muta, H. & Yamanaka, S. Chalcopyrite CuGaTe2: a high-efficiency bulk thermoelectric material. Adv. Mater. 24, 3622–3626 (2012).
Ibáñez, M., Cadavid, D., Anselmi-Tamburini, U., Zamani, R., Gorsse, S., Li, W., López, A. M., Morante, J. R., Arbiol, J. & Cabot, A. Colloidal synthesis and thermoelectric properties of Cu2SnSe3 nanocrystals. J. Mater. Chem. A 1, 1421–1426 (2013).
Skoug, E. J., Cain, J. D. & Morelli, D. T. High thermoelectric figure of merit in the Cu3SbSe4-Cu3SbS4 solid solution. Appl. Phys. Lett. 98, 261911 (2011).
Li, Y., Zhang, T., Qin, Y., Day, T., Jeffrey Snyder, G., Shi, X. & Chen, L. Thermoelectric transport properties of diamond-like Cu1−xFe1+xS2 tetrahedral compounds. J. Appl. Phys. 116, 203705 (2014).
Ang, R., Khan, A. U., Tsujii, N., Takai, K., Nakamura, R. & Mori, T. Thermoelectricity generation and electron–magnon scattering in a natural chalcopyrite mineral from a deep-sea hydrothermal vent. Angew. Chem. Int. Ed. 54, 12909–12913 (2015).
Li, J., Tan, Q. & Li, J.-F. Synthesis and property evaluation of CuFeS2−x as earth-abundant and environmentally-friendly thermoelectric materials. J. Alloy Compd. 551, 143–149 (2013).
Berthebaud, D., Lebedev, O. I. & Maignan, A. Thermoelectric properties of n-type cobalt doped chalcopyrite Cu1−xCoxFeS2 and p-type eskebornite CuFeSe2 . J. Materiomics 1, 68–74 (2015).
Sato, K., Harada, Y., Taguchi, M., Shin, S. & Fujimori, A. Characterization of Fe 3d states in CuFeS2 by resonant X-ray emission spectroscopy. Phys. Stat. Solid. A 206, 1096–1100 (2009).
Xie, H., Su, X., Zheng, G., Zhu, T., Yin, K., Yan, Y., Uher, C., Kanatzidis, M. G. & Tang, X. The role of Zn in chalcopyrite CuFeS2: enhanced thermoelectric properties of Cu1-xZnxFeS2 with in situ nanoprecipitates. Adv. Energy Mater. 7, 1601299 (2016).
Xie, H., Su, X., Zheng, G., Yan, Y., Liu, W., Tang, H., Kanatzidis, M. G., Uher, C. & Tang, X. Nonmagnetic in substituted CuFe1–xInxS2 solid solution thermoelectric. J. Phys. Chem. C 120, 27895–27902 (2016).
Carr, W. D. & Morelli, D. T. The thermoelectric properties and solubility limit of CuFeS2(1−x)Se2x . J. Electron Mater 45, 1346–1350 (2015).
Tsujii, N., Mori, T. & Isoda, Y. Phase stability and thermoelectric properties of CuFeS2-based magnetic semiconductor. J. Electron Mater. 43, 2371–2375 (2014).
Tsujii, N. & Mori, T. High thermoelectric power factor in a carrier-doped magnetic semiconductor CuFeS2 . Appl. Phys. Express 6, 043001 (2013).
Tsujii, N. Possible enhancement of thermoelectric properties by use of a magnetic semiconductor: carrier-doped chalcopyrite Cu1−xFe1+xS2 . J. Electron Mater 42, 1974–1977 (2013).
Liang, D., Ma, R., Jiao, S., Pang, G. & Feng, S. A facile synthetic approach for copper iron sulfide nanocrystals with enhanced thermoelectric performance. Nanoscale 4, 6265–6268 (2012).
Su, X., Fu, F., Yan, Y., Zheng, G., Liang, T., Zhang, Q., Cheng, X., Yang, D., Chi, H., Tang, X., Zhang, Q. & Uher, C. Self-propagating high-temperature synthesis for compound thermoelectrics and new criterion for combustion processing. Nat. Commun. 5, 4908 (2014).
Zheng, G., Su, X., Liang, T., Lu, Q., Yan, Y., Uher, C. & Tang, X. High thermoelectric performance of mechanically robust n-type Bi2Te3−xSex prepared by combustion synthesis. J. Mater. Chem. A 3, 6603–6613 (2015).
Liang, T., Su, X., Yan, Y., Zheng, G., Zhang, Q., Chi, H., Tang, X. & Uher, C. Ultra-fast synthesis and thermoelectric properties of Te doped skutterudites. J. Mater Chem. A 2, 17914–17918 (2014).
She, X., Su, X., Du, H., Liang, T., Zheng, G., Yan, Y., Akram, R., Uher, C. & Tang, X. High thermoelectric performance of higher manganese silicides prepared by ultra-fast thermal explosion. J. Mater Chem. C 3, 12116–12122 (2015).
Bergman, D. J. & Fel, L. G. Enhancement of thermoelectric power factor in composite thermoelectrics. J. Appl. Phys. 85, 8205–8216 (1999).
Bergman, D. J. & Levy, O. Thermoelectric properties of a composite medium. J. Appl. Phys. 70, 6821–6833 (1991).
Padilla, R., Rodriguez, M. & Ruiz, M. C. Sulfidation of chalcopyrite with elemental sulfur. Metall. Mater. Trans. B. 34, 15–23 (2003).
Zhao, L. D., Lo, S. H., He, J., Li, H., Biswas, K., Androulakis, J., Wu, C. I., Hogan, T. P., Chung, D. Y., Dravid, V. P. & Kanatzidis, M. G. High performance thermoelectrics from earth-abundant materials: enhanced figure of merit in PbS by second phase nanostructures. J. Am. Chem. Soc. 133, 20476–20487 (2011).
Liu, W.-S., Zhang, Q., Lan, Y., Chen, S., Yan, X., Zhang, Q., Wang, H., Wang, D., Chen, G. & Ren, Z. Thermoelectric property studies on Cu-doped n-type CuxBi2Te2.7Se0.3 nanocomposites. Adv. Energy Mater. 1, 577–587 (2011).
Xie, W. J., He, J., Zhu, S., Su, X. L., Wang, S. Y., Holgate, T., Graff, J. W., Ponnambalam, V., Poon, S. J. & Tang, X. F. Simultaneously optimizing the independent thermoelectric properties in (Ti,Zr,Hf)(Co,Ni)Sb alloy by in situ forming InSb nanoinclusions. Acta Mater. 58, 4705–4713 (2010).
Pei, Y. L., Wu, H., Wu, D., Zheng, F. & He, J. High thermoelectric performance realized in a BiCuSeO system by improving carrier mobility through 3D modulation doping. J. Am. Chem. Soc. 136, 13902–13908 (2014).
Yu, B., Zebarjadi, M., Wang, H., Lukas, K., Wang, H., Wang, D., Opeil, C., Dresselhaus, M., Chen, G. & Ren, Z. Enhancement of thermoelectric properties by modulation-doping in silicon germanium alloy nanocomposites. Nano Lett. 12, 2077–2082 (2012).
Zebarjadi, M., Joshi, G., Zhu, G., Yu, B., Minnich, A., Lan, Y., Wang, X., Dresselhaus, M., Ren, Z. & Chen, G. Power factor enhancement by modulation doping in bulk nanocomposites. Nano Lett. 11, 2225–2230 (2011).
Acknowledgements
We acknowledge support from the National Basic Research Program of China (973 program) under project 2013CB632502, the Natural Science Foundation of China (Grant Number 51402222, 51172174, 51521001 and 51632006) and the 111 Project of China (Grant Number B07040). CU and XT also acknowledge support provided by the US-China CERC-CVC program under Award Number DE-PI0000012.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the NPG Asia Materials website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Xie, H., Su, X., Yan, Y. et al. Thermoelectric performance of CuFeS2+2x composites prepared by rapid thermal explosion. NPG Asia Mater 9, e390 (2017). https://doi.org/10.1038/am.2017.80
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/am.2017.80
This article is cited by
-
63,65Cu NMR and EPR Study of Doped Chalcopyrite Cu1-XPdXFeS2 Compounds
Applied Magnetic Resonance (2021)
-
Achieving superior performance in thermoelectric Bi0.4Sb1.6Te3.72 by enhancing texture and inducing high-density line defects
Science China Materials (2021)
-
Optimizing thermoelectric properties of BiSe through Cu additive enhanced effective mass and phonon scattering
Rare Metals (2020)
-
Structural and Thermoelectric Properties of Solid–Liquid In4Se3-In Composite
Journal of Electronic Materials (2019)
