
Abstract
An association between the rare condition of transient neonatal diabetes mellitus and either uniparental disomy for chromosome 6 or dup(6)(q22q23) raised the assumption that in this location on chromosome 6 there is an imprinted gene. We diagnosed diabetes that developed in a baby girl immediately after birth and resolved after 7 weeks of insulin treatment. Due to some minor dysmorphic features, we investigated her karyotype and identified inv-dup(6)(q22q23). The duplication spans at least 10 cM including the DNA sites DS270, S314, S1684 and S310. This case further supports the assumption that an imprinted gene exists on chromosome 6q22–23.
Similar content being viewed by others
Introduction
Neonatal diabetes mellitus (NDM) is a very rare entity, with an estimated incidence of 1 in 400,000 neonates [1]. Patients with NDM require insulin therapy to maintain euglycemia. In about half of the neonates, the diabetes is transient (TNDM) and resolves at a median age of 3 months, while the rest of the patients have a permanent form of diabetes. In addition, in a significant number of patients with TNDM, diabetes type 2 (IDDM) reappears later in life. The association between TNDM and either paternal isodisomy or duplication of chromosome 6q22–23 raised the possibility of an imprinted gene in this location. We report on a child with TNDM with a de novo invdup(6)(q22q23); the duplicated segment was limited to the segment between the DNA loci D6S308 and D6S1684.
Patient Report
The proband is a female, the first child of Ashkenazi Jewish parents aged 25 and 26 years. She was born at 36 weeks of gestation and weighed 1,660 g (small for gestational age). Almost immediately after birth persistent hyperglycemia was documented and the diagnosis of NDM was made. She was successfully treated with insulin injections till the age of 7 weeks to maintain euglycemia. Because of her relatively coarse facial features and a protruding tongue, a cytogenetic analysis was performed. At the age of 1 year she weighed 8.5 kg; upon evaluation at the age of 2 years, her mental and motor development were normal, physical examination was normal except for isolated macroglossia.
Materials and Methods
Cytogenetic Analysis
Peripheral blood lymphocytes were cultured according to standard techniques [2]. For high-resolution banding analysis, MTX was applied to the cultures. G-banding was performed according to the standard techniques [3]. FISH analysis was performed using chromosome 6 painting probe (Oncor, Gaithersburg, Md., USA) according to the manufacturer’s instructions.
DNA Analysis
Genomic DNA was extracted according to standard techniques. Short tandem repeat (STR) sequence polymorphisms were analyzed as previously described [4]. The following (CA)n dinucleotide repeat polymorphisms were used: D6S292, D6S314, D6S1684, D6S310, D6S308, D6S311, D6S1577, D6S305, the sequences of oligonucleotide primers were retrieved from on-line Genome Data Base (GDB).
Results
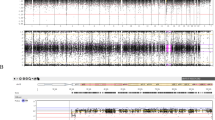
Cytogenetic analysis on the patient’s lymphocytes revealed aberrant chromosome 6 with extra material on chromosome 6q (fig. 1), we suspected that it was a duplication of the 6q segment. FISH, using chromosome 6 painting probe painted the extra material on chromosome 6. According to the G-banding pattern, we interpreted the karyotype as 46,XX,dup(6)(q16q21). The parental karyotypes were normal.

Partial karyotype of the patient and schematic presentation of the evolution of the abnormal chromosome.
In order to better define the duplicated region and its origin, we analyzed the segregation of DNA polymorphic markers (table 1). Fully informative markers were those in which the parents did not share a common allele and both were heterozygotes, such as D6S1684. The informative markers indicated that the duplicated chromosome was of paternal origin and that the duplication arose from a single chromosome. Namely, the father contributed two doses of the same allele as could be seen from the relative intensity of the relevant PCR products. The duplication spans at least 10 cM including the markers D6S1684, D6S310 and D6S308. The DNA marker D6S311 which lies 3 cM telomeric to D6S308 was not duplicated (fig. 2). The DNA markers on the duplicated segments with definite map position pointed to duplication of DNA markers from 6q22–23 segment. We thus re-evaluated the karyotype according to the molecular results and the G-banding pattern and reached the conclusion that the duplication was a result of inversion duplication of the segment 6q22–23 (fig. 1).

Genetic map of the duplicated segment on 6q (the order of the DNA markers is based on the on-line Chromosome 6 version v8c7 Integrated Marker Map.
Discussion
In 1994, Abramowicz et al. [5] reported on a child with methylmalonic acidemia and NDM. A paternal isodiso-my (UPD) of chromosome 6 was demonstrated which caused homozygotization of a mutated paternal allele of the gene MUT leading to methylmalonic acidemia. The authors speculated on the existence of a gene on chromosome 6 which is involved in the pathogenesis of NDM. Since this first publication, there were reports on 3 children with TNDM associated with paternal UPD for chromosome 6 [7] which support the hypothesis of an imprinted gene on chromosome 6. Another association of a chromosome 6 anomaly and TNDM was found in patients with duplication of 6q23 of paternal origin [8, 9]. These observations led Temple et al. [10] to systematically search for chromosome 6 abnormalities in 10 patients of unrelated families with NDM. In one family (family A), they found familial duplication on (6)(q22q23) that caused TNDM upon paternal transmission, in another familial case with TNDM (family B), they demonstrated a linkage to markers from the same region of chromosome 6. The mode of inheritance in these two families further supports the existence of an imprinted gene on chromosome 6q22–23. In the present report an additional child with TNDM is described in association with dup(6)(q22 q23) of paternal origin and thus adds another case in support of the concept of an imprinted gene on chromosome 6. The duplication spans the same region as that of family A of Temple et al. [10]. In our patient the duplication involves one of the paternal homologues of chromosome 6 which, according to our interpretation, was a result of inversion duplication of 6q22–q23. Whether this region is a hot spot for rearrangement due to homologous repetitive sequences flanking the putative imprinted gene needs further study.
While general clinical data of the prognosis of children with TNDM are available, there are still too few patients in whom the NDM was transient and caused by UPD6 or duplication of (6)(q22q23) of the paternal chromosome to conclude what is their long-term prognosis. Of interest are the patients with UPD6 [6], one that developed IDDM at the age of 13 and one who remained euglycemic at the age of 3 years, while in the family reported by Temple et al. [10], 4 sibs remained asymptomatic at the ages 6, 32, 30 and 25 years after their TNDM event. Identifying other previously reported cases of TNDM and performing comprehensive cytogenetic studies might be an avenue to building an adequate data base and evaluating the exact prognosis.
References
Shield JP, Gardner RJ, Wadsworth EJ, White-ford EL, James RS, Robinson DO, Baum JD, Temple IK: Aetiology and genetic basis of neonatal diabetes. Arch Dis Child Fetal Neonatal Ed 1997;76:F39–42.
Gosden CM, Davidson C, Robertson M: Lymphocyte culture; in Rooney ED, Czepulkowski BH (eds): Human Cytogenetics, Constitutional Analysis. A Practical Approach, ed 2. Washington, IRL Press, 1992, vol 1, pp 31–54.
Benn PA, Perle MA: Chromosome staining and banding techniques; in Rooney DE, Czepulkowski BH (eds): Human Cytogenetics, Constitutional Analysis. A Practical Approach, ed 2. Washington, IRL Press, 1992, vol 1, pp 91–118.
Lerer I, Meiner V, Pashut-Lavon I, Abeliovich D: Molecular diagnosis of Prader-Willi syndrome: Parent-of-origin dependent methyl-ation sites and non-isotopic detection of (CA)n dinucleotide repeat polymorphisms. Am J Med Genet 1994;52:79–84.
Abramowicz MJ, Andrien M, Dupont E, Dor-chy H, Parma J, Duprez J, Ledley FD, Cour-tens W, Vamos E: Isodisomy of chromosome 6 in a newborn with methylmalonic acidemia and agenesis of pancreatic beta cells causing diabetes mellitus. J Clin Invest 1994;94:418–421.
Temple KI, James RS, Crolla JA, Sitch FL, Jacobs PA, Howell MW, Betts P, Baum DJ, Shield JPH: An imprinted gene(s) for diabetes (?). Nat Genet 1996;9:110–112.
Whiteford ML, Narenda A, White MP, Cooke A, Wilkinson AG, Robertson KJ, Tolmie JL: Paternal uniparental disomy for chromosome 6 causing transient neonatal diabetes. J Med Genet 1997;34:167–168.
Pivnik EK, Qumsiyeh MB, Tharapel AT, Sum-mitt JB, Wilroy RS: Partial duplication of the long arm of chromosome 6: A clinically recognizable syndrome. J Med Genet 1990;27:523–526.
Ziel B, Zneimer SM, Bachman R: Partial trisomy of chromosome 6q, an interstitial duplication of the long arm (abstract 741). Am J Hum Genet 1995;53(4, suppl):A131.
Temple KI, Gardner RJ, Robinson DO, Kibir-ige MS, Ferguson AW, Baum JD, Barber JCK, James RS, Shield JPH: Further evidence for an imprinted gene for neonatal diabetes localised to chromosome 9q22–q23. Hum Mol Genet 1995;5:1117–1124.
Acknowledgment
We wish to thank Karen Temple, FRCP, for sharing with us the information regarding the polymorphic loci within the 6q duplication in TNDM.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Arthur, E.I., Zlotogora, J., Lerer, I. et al. Transient Neonatal Diabetes mellitus in a Child with invdup(6)(q22q23) of Paternal Origin. Eur J Hum Genet 5, 417–419 (1997). https://doi.org/10.1007/BF03405952
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/BF03405952
