
Abstract
Branchio-oto-renal (BOR) syndrome is an autosomal dominant disease characterized by varying combinations of branchial, otic and renal anomalies. By positional cloning, a candidate gene, EYA1, homologous to the drosophila eyes absent gene, has recently been identified at 8q13.3 and shown to underlie this syndrome. The name branchio-oto (BO) syndrome has been used to describe a similar combination of branchial and otic anomalies, without the association of renal anomalies. Whether BOR and BO syndromes involve the same gene was unknown. To address this question, we analyzed two large independent families for which each of the 8 affected members present exclusively with BO syndrome. In both families, linkage analysis mapped the causative gene to the same chromosomal region as EYA1. A search for mutations in 9 of the EYA1 coding exons identified a 2-bp insertion segregating in one family and an 8-bp deletion segregating in the other. These results demonstrate that EYA1 also underlies BO syndrome, and that BOR and BO syndromes are allelic defects of this gene.
Similar content being viewed by others
Introduction
The name branchio-oto-renal (BOR) syndrome was first used in 1975 [1], to refer to a syndromic form of deafness associating branchial anomalies, i.e. clefts, fistulas, or cysts, otic anomalies affecting the outer, middle and/or inner ear and renal anomalies ranging from mild dysplasia to bilateral aplasia. BOR syndrome has an autosomal dominant mode of inheritance with variable expressivity and incomplete penetrance. Deafness can range from mild to severe, and this syndrome has been estimated to account for 2% of profoundly deaf children [2]. The identification of an 8q chromosomal rearrangement in a patient presenting with tricho-rhino-phalangeal, branchial and otic anomalies [3] had focused the linkage analysis of BOR-affected families to this chromosomal region [4]. The gene responsible for BOR syndrome was subsequently mapped between the markers D8S543 and D8S286, a distance of 7 cM [5]. Further characterization of the aforementioned chromosomal rearrangement led to the identification of an associated deletion [6, 7] that allowed refinement of the gene interval to 470–650 kb [7]. Recently, using a positional cloning strategy, we identified and cloned a human homologue of the drosophila eyes absent gene [8], EYA1, mapping within the gene interval [9]. The identification of seven different mutations within EYA1 in BOR-affected patients established that this gene underlies BOR syndrome [9]. Other syndromes, branchio-oto (BO), branchio-oto-ureteral (BOU), comprising abnormalities of the branchial arches and deafness have been described [for a review, see 10]. BO-affected individuals present with the same branchial and otic anomalies as those seen in BOR-affected patients but lack renal anomalies. Due to the existence of families made up exclusively of BO-affected individuals, BO syndrome was initially considered to arise from either a different mutation in the gene responsible for BOR syndrome or a defect in a different gene [1]. Subsequent reports describing individuals within the same family affected with either BOR or BO syndrome led to the conclusion that, at least in some cases, the same mutation underlies both diseases [11]. However, to date, only a few BOR-affected families have been reported to carry mutations in EYA1 and no BOR/BO-affected families have been tested [9]. Moreover, even though no genetic heterogeneity has been reported for BOR syndrome, this possibility cannot be eliminated. To address the molecular basis of BO syndrome, BO-affected families were collected. Preliminary linkage analysis of one of these families argued that the causative gene mapped to the same interval as the gene responsible for BOR syndrome [5]. We report here on the linkage analysis of the other large family that confirms this earlier localization. In addition, the identification of mutations in the coding exons of EYA1 in both families demonstrates that the same gene underlies both BOR and BO syndrome.
Materials and Methods
Linkage Analysis
Genomic DNA was prepared from peripheral blood lymphocytes using standard procedures. A set of 4 microsatellite markers mapping proximal (D8S543) and distal (D8S530, D8S279, D8S286) to the translocation-associated deletion [7] were chosen for linkage analysis. Amplification and gel analysis of the microsatellite repeats were performed as described [5]. Two-point lodscores between the BO locus and each of the microsatellite markers, were calculated using the programs MLINK and ILINK of the LINKAGE package (version 5.2) [12]. The pairwise lodscores were estimated using a penetrance value of 100% and the Généthon derived allele frequencies [13].
Mutation Characterization
Eight of the C-terminal coding exons of EYA1, exon z and exons A to G, encoding the protein sequence from amino acid position 241 to 533, were amplified and sequenced using intronic primers [9]. Amplification and subcloning of PCR products into a HincII digested Ml3 vector were carried out as previously described [14]. By genomic sequencing, an exon, exon z′, encoding the protein sequence from amino acid position 59 to 107, was identified. Exon z′ was amplified and sequenced using the following intronic primers:
exz′.F | 5′GTGATGTGGTTGTTAATCGGT3′ |
|---|---|
exz′.R | 5′ACACAGAAGGTGACAACACGT3′ |
PCR amplification for the exon z′ was carried out as previously described [14].
Results
The Gene for BO Syndrome Maps to the Same Region as EYA1
To ensure the correct diagnosis of BO syndrome, affected and unaffected individuals from families A and B underwent clinical, biological and radiological examinations. All the affected individuals examined (5 from family A and 7 from family B) presented with branchial anomalies consisting of branchial cervical fistulas, situated mainly at the level of third branchial arch in family A and at the level of second branchial arch in family B. The outer ear anomalies consisted of a cup-shaped pinna (3 individuals from family A and 4 from family B) and a preauricular pit (4 individuals from family A and 5 from family B). Audiometric testing was performed at frequencies of 125, 250, 1,000, 2,000, 4,000 and 8,000 Hz for pure-tone air conduction and 250, 500, 1,000, 2,000 and 4,000 Hz for bone conduction. A pure sensorineural deafness, with a mean hearing loss of 45 dB HL (decibels hearing level), was noted in 1 individual from family A and in 2 from family B. A mixed-type deafness (sensorineural and conductive), with a mean hearing loss of 60 dB HL, was observed in 4 individuals from both families A and B. A pure conductive deafness, with a mean hearing loss of 40 dB HL (associated with a sensorineural notch centered on frequency 2,000 Hz), was observed in only one individual (family B). No evidence of renal anomalies was obtained by clinical examination, biological analysis of renal function (uremia and creatininemia) nor by high-resolution renal ultrasonography.
Previous linkage analysis of family A resulted in a maximum lodscore of 1.5 for marker D8S279 (at θ = 0) [5]. Linkage analysis using the polymorphic markers flanking the aforementioned translocation-associated deletion (see Materials and Methods) was performed for 7 affected individuals from family B, as well as for unaffected members. A maximum lodscore of 2.67 for marker D8S279 (at θ = 0) was obtained for family B. In neither of the two families did a recombination event occur between this marker and the gene interval in affected or unaffected individuals (fig. 1). Previous results for unaffected individual 16 [VIIIP in 5] from family A have shown that a recombination event between the marker D8S543 and the disease locus had occurred, thus localizing the gene responsible for BO syndrome distal to that marker [5]. In individual 13 from family B, a recombination event has occurred between the marker D8S286 and the disease locus, thus mapping the gene proximal to this marker. These two recombination events map the causative gene to a 7-cM interval flanked by D8S543 and D8S286; this interval contains EYA1.

Pedigrees of families affected with BO syndrome, showing haplotypes for marker D8S279 and flanking recombinant markers D8S543 and D8S286. Filled symbols represent patients exhibiting deafness and branchial anomalies, empty symbols correspond to unaffected individuals.
EYA1 Mutations in BO-Affected Individuals
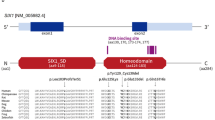
A search for possible DNA rearrangements within EYA1 in BO-affected individuals was performed by Southern blot analysis. Probes corresponding to the EYA1 cDNA were hybridized to Southern blots containing TaqI, MspI and EcoRI digested DNA from individuals of both families. No DNA rearrangements were detected (data not shown). A search for mutations was subsequently performed by sequencing. We had previously defined the limits of exons A to G, and exon z, which encode part of the C-terminus of the protein (amino acid position 241–533) [9]. Additional genomic sequences obtained allowed us to characterize another exon (encoding a 49 amino acid fragment, position 59 to 107, see Materials and Methods) hereafter referred to as exon z′. Sequence analysis of these 9 exons in both families identified two mutations (fig. 2). In each of the affected individuals from family A, a mutation consisting of a 2-bp (GT) insertion in exon A, at position 870, was observed. Within family B, each affected member carried an 8-bp deletion in exon z′, at position 297. The two mutations result in a frameshift of the coding phase leading to a premature stop codon. None of the unaffected members of either family carried these mutations.

EYA1 mutations in exon A and exon z′.
Discussion
Recently, EYA1 has been demonstrated to underlie BOR syndrome [9]. Analysis of the expression pattern of the orthologous murine gene, Eya1, has shown that this gene is expressed early in the developing ear (middle and inner ear), as well as in the metanephric mesenchymal cells just as they begin to condense due to an induction by the dividing ureteric branches. These results are consistent with the clinical and paraclinical features of BOR syndrome which are indicative of a developmental defect taking place between the 4th and the 10th week of embryogenesis [9]. The nuclear localization of the drosophila eyes absent gene product [8] suggests that EYA1 may encode a developmental transcription factor. However, the nuclear localization of this 559 amino acid predicted protein has yet to be demonstrated.
The results presented here show that mutations in EYA1 can be responsible for BO syndrome. The two mutations described above have been transmitted over 3–4 generations with complete penetrance and do not result in renal anomalies in any of the 12 BO-affected individuals examined. Three main hypotheses could account for mutations in EYA1 being responsible for either BOR or BO syndrome. First, the type of mutations previously detected in BOR-affected patients suggests that this disease results from haploinsufficiency, thus indicating that the normal activity of EYA1 is close to the threshold level required for the appearence of clinical defects. One can predict that a minimal increase in protein concentration, relative to the critical level, can prevent the appearance of some of the symptoms. In the context of this hypothesis, renal development would be less sensitive to a decrease in EYA1 concentration than would the development of the ear and branchial arches. Second, a modifier gene could modulate the phenotypic expression of EYA1 mutation. Third, different types of mutations within EYA1 could account for both BOR and BO syndromes. The first hypothesis does not explain the fact that all affected individuals from families A and B have the same phenotypic features. The possible existence of a modifier gene cannot be ruled out, but such a gene would have to cosegregate with EYA1 and thus lie in close proximity to this gene. The first two hypotheses are well suited for families in which some members are affected by BOR syndrome and some by BO syndrome. The last hypothesis seems to be the most attractive explanation for families in which members are exclusively affected by BO syndrome. The two observed mutations reported here lead to a frameshift and are thus expected to be as deleterious as the 7 previously detected mutations in BOR-affected patients [9]. Nevertheless, no mutation in the two exons (A and z′) has been found in the BOR-affected patients tested to date. Accordingly, it is tempting to propose that these two exons are spliced out in at least some EYA1 kidney transcripts. Examination of the 5′ and 3′ exon boundaries indicate that exon A can be individually spliced out without modification of the reading frame, while exon z′ would have to be spliced out with other exon(s) in order to maintain the ORF. Exon A encodes part of the region sharing high homology with the drosophila eyes absent gene product, named eyaHR, a region shared by the two other identified members of the EYA family, EYA2 and EYA3 [9]. Exon z′ encodes part of the N-terminal region, which is not highly conserved. We have recently obtained indications for the existence of various trancripts derived from EYA1 [unpubl. data]. Even if, for the time being, these alternatively spliced products do not involve exons A and z′, the present results would incite a search for such transcripts during the period of kidney morphogenesis which is disrupted in BOR-affected embryos. Finally, this work suggests that EYA1 is a good candidate gene for other syndromes associating branchial and otic anomalies, such as BOU syndrome.
References
Melnick M, Bixler D, Nance W, Silk K, Yune H: Familial branchio-oto-renal dysplasia: A new addition to the branchial arch syndromes. Clin Genet 1976;9:25–34.
Fraser FC, Sproule JR, Hatal F: Frequency of the branchio-oto-renal (BOR) syndrome in children with profound hearing loss. Am J Med Genet 1980;7:341–349.
Haan EA, Hull YJ, White S, Cockington R, Charlton P, Callen DF: Tricho-rhino-phalangeal and branchio-oto syndromes in a family with an inherited rearrangement of chromosome 8q. Am J Med Genet 1989;32:490–494.
Kumar S, Kimberling WJ, Kenyon JB, Smith RJH, Marres HAM, Cremers CWRJ: Autosomal dominant branchio-oto-renal syndrome-localization of a disease gene to chromosome 8q by linkage in a Dutch family. Hum Mol Genet 1992;1:491–495.
Vincent C, Kalatzis V, Compain S, Levilliers J, Slim R, Graia F, Pereira MdL, Nivelon A, Croquette MF, Lacombe D, Vigneron J, Helias J, Broyer M, Callen DF, Haan EA, Weissenbach J, Lacroix B, Bellané-Chantelot C, Le Paslier D, Cohen D, Petit C: A proposed new contiguous syndrome on 8q consists of branchio-otorenal (BOR) syndrome, Duane syndrome, a dominant form of hydrocephalus and trapese aplasia; implications for the mapping of the BOR gene. Hum Mol Genet 1994;3:1859–1866.
Gu JZ, Wagner MJ, Haan EA, Wells DE: Detection of a megabase deletion in a patient with branchio-oto-renal syndrome (BOR) and tricho-rhino-phalangeal syndrome (TRPS): Implications for mapping and cloning the BOR gene. Genomics 1996;31:201–206.
Kalatzis V, Abdelhak S, Compain S, Vincent C, Petit C: Characterization of a translocation-associated deletion defines the candidate region for the gene responsible for branchio-otorenal syndrome. Genomics 1996;34:422–425.
Bonini NM, Leiserson WM, Benzer S: The eyes absent gene: Genetic control of cell survival and differentiation in the developing drosophila eye. Cell 1993;72:379–395.
Abdelhak S, Kalatzis V, Heilig R, Compain S, Samson D, Vincent C, Weil D, Cruaud C, Sahly I, Leibovici M, Bitner-Glindzicz M, Francis M, Lacombe D, Vigneron J, Charachon R, Boven K, Bedbeder P, Van Regemorter N, Weissenbach J, Petit C: A human homologue of the Drosophila eyes absent gene underlies branchio-oto-renal (BOR) syndrome and identifies a novel gene family. Nature Genet 1997; 15:157–164.
Gorlin RJ, Toriello HV, Cohen MM: Hereditary hearing loss and its syndromes; in Oxford Monographs on Medical Genetics. New York, Oxford University Press, 1995, vol 28, p 91.
Heimler A, Lieber E: Branchio-oto-renal syndrome: reduced penetrance and variable expressivity in four generations of a large kindred. Am J Hum Genet 1986;2:241–252.
Lathrop GM, Lalouel JM, Julier C, Ott J: Multilocus linkage analysis in humans: Detection of linkage and estimation of recombination. Am J Hum Genet 1985;37:482–498.
Dib C, Fauré S, Fizames C, Samson D, Drouot N, Vignal A, Millaseau P, Marc S, Hazan J, Seboun E, Lathrop M, Gyapay G, Morissette J, Weissenbach J: A comprehensive genetic map of the human genome based on 5,264 microsatellites. Nature 1996;380:152–154.
Weil D, Blanchard S, Kaplan J, Guilford P, Gibson F, Walsh J, Mburu P, Varela A, Levilliers J, Weston MD, Kelley PM, Kimberling WJ, Wagenaar M, Levi-Acobas F, Larget-Piet D, Munnich A, Steel KP, Brown SDM, Petit C: Defective myosin VIIA gene responsible for Usher syndrome type 1B. Nature 1995;374: 60–61.
Acknowledgements
V. Kalatzis is supported by the Fondation pour la Recherche Médicale, S. Abdelhak by the Association Française contre les Myopathies (AFM) and H. Chaïb by the Association Claude Bernard. This work was supported by grants from the AFM (4402 MG-1996) and the Commission of the European Community (PL95-1324).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Vincent, C., Kalatzis, V., Abdelhak, S. et al. BOR and BO Syndromes Are Allelic Defects of EYA1. Eur J Hum Genet 5, 242–246 (1997). https://doi.org/10.1007/BF03405924
Received:
Revised:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/BF03405924
