
Abstract
Therapeutic cloning, whereby embryonic stem cells (ESCs) are derived from nuclear transfer (NT) embryos, may play a major role in the new era of regenerative medicine. In this study we established forty nuclear transfer-ESC (NT-ESC) lines that were derived from NT embryos of different donor cell types or passages. We found that NT-ESCs were capable of forming embryoid bodies. In addition, NT-ESCs expressed pluripotency stem cell markers in vitro and could differentiate into embryonic tissues in vivo. NT embryos from early passage R1 donor cells were able to form full term developed pups, whereas those from late passage R1 ES donor cells lost the potential for reprogramming that is essential for live birth. We subsequently established sequential NT-R1-ESC lines that were developed from NT blastocyst of late passage R1 ESC donors. However, these NT-R1-ESC lines, when used as nuclear transfer donors at their early passages, failed to result in live pups. This indicates that the therapeutic cloning process using sequential NT-ESCs may not rescue the developmental deficiencies that resided in previous donor generations.
Similar content being viewed by others

Introduction
Since the generation of “Dolly,” a wide range of mammals have been cloned by somatic cell nuclear transfer (SCNT), and successful genomic reprogramming has been shown to occur in the donor cell nucleus 1. More recently, customized pluripotent embryonic stem cells (ESC) have been derived from nuclear transfer embryos by therapeutic cloning in several labs 2, 3. The ES cell lines generated were shown to be able to differentiate into all three embryonic germ layers both in vitro and in vivo, as well as form germlines leading to mature spermatozoa and oocytes 4, 5, 6, 7, 8, 9, 10. Furthermore, the nuclear transfer ESCs (NT-ESCs) were shown to differentiate into dopaminergic neurons which could ameliorate symptoms in a mouse Parkinson disease model 11. A number of reports have shown the potential usefulness of therapeutic cloning using ES cells derived from cloned animals (NT-ESCs) in regenerative medicine to rescue immune deficient or degenerative phenotypes 12, 13, 14. Other evidence suggested that NT-derived ES cells in mouse have similar developmental potential as fertilization-derived ES cells, and appear to be indistinguishable from their fertilization-derived counterparts on a molecular level 2, 3.
Despite the potential usefulness, nuclear transfer (NT) remains an inefficient process. Most reconstructed embryos failed to develop to term; and when born alive, cloned animals usually display a neonatal phenotype resembling large offspring syndrome with multiple systematic abnormalities as well as dysfunctional placentas 15, 16. In addition, genetic or epigenetic defects such as DNA rearrangements were frequently found in mature central nervous system neurons 17, and in stem cells with prolonged cell culture exposure 18. Several reports showed that defective cloned embryos can still supply ESCs from which specific cell types could be derived 19. Thus, numerous safety issues must be examined and solved before therapeutic cloning can be applied to cell therapy and preclinical trials for disease treatment in regenerative medicine.
In this study, we used somatic cells and the routinely-used R1 ES cells in NT to examine the reprogramming capability associated with donors of various source and passages, and evaluated the developmental effect of therapeutic cloning using NT-ESCs from these cloned embryos. Forty ES cell lines were generated from these cloned embryos in the process, which may provide additional resources for future studies.
Materials and Methods
Mature oocytes collection and donor cell preparation
B6D2 (C57BL/6×DBA) F1 females (8∼10 weeks old) were superovulated and cumulus-oocyte complexes (COC) were collected at 13∼14 h after hCG injection. Cumulus cells were removed by treatment with 300U/ml hyaluronidase (ICN Pharmaceuticals, Costa Mesa, CA), and oocytes were cultured in CZB medium before micromanipulation.
Somatic cell (cumulus cell) used for nuclear donors were washed 2∼3 times in drops of CZB-Hepes and transferred directly to micromanipulation chamber. Synchronized embryonic stem cells were used for nuclear transfer, which were in metaphase as previously described 20.
Nuclear transfer and embryo transfer
Nuclear transfer was performed as previously described 21. Donor nuclei or chromosomes were removed from donor cells (cumulus nucleus or metaphase ES nucleus) by gently aspirating in and out of the injection pipette, and then injecting into recipient oocytes. The meiotic metaphase plate was removed while withdrawing the pipette from cytoplasm after injection. The donor cells were cumulus cells, ntES1 (established in our lab) and R1 (gift from Prof. Nagy's laboratory). Twenty to thirty oocytes were placed into the chamber containing 1 ml Hepes-CZB medium with 5 μg/ml cytochalasin B (CB) covered by mineral oil. Pipettes with an internal diameter of 8∼12 μm were used for the injection of the donor nucleus using a Piezo-electric device (P150, PrimeTech Japan). 1∼2 h after injection, the reconstructed embryos from cumulus cells were activated by a 5 h incubation in calcium-free CZB supplemented with 10 mM SrCl2 and 5 μg/ml Cytochalasin B (CB) before being extensively washed and cultured in CZB medium at 37°C and 5% CO2 for 4 d; Reconstructed embryos from ES cells were activated by only 3-h culture in 10 mM SrCl2 CZB medium without CB and calcium. In vivo fertilized oocytes were cultured as a control. CD-1 females were used as embryo recipients. Nuclear transfer embryos at the two- or four-cell stage were transferred to the ampullae of oviducts of females on half a day after mating. Transferred embryos were recovered by Caesarian section at day 19.5. Lactating CD-1 foster mothers were used to raise live pups.
Stem cells culture and establishment of NT-ESC lines
Stem cells were cultured as described 22, 23 with DMEM/F12 (1:1, Gibco No.11320-033) plus 20% knockout serum (Gibco No.10828), LIF1000U (leukaemia inhibitory factor, Chemicon, ESG1107), 2 mM glutamine (Sigma, No.G8540), 1 mM sodium pyruvate (Sigma, No.L1375), 0.1 mM β-mercaptoethanol (Sigma, No.M6250), 0.1 mM non-essential amino acid (NEAA, Gibco No.11140-050).
Cloned blastocysts with zona pollucida removed were placed in the 4-well dish precoated with mouse embryonic fibroblasts, proliferating outgrowths were dissociated using manual pipetting or 0.25% trypsin treatment, and then re-plated on fibroblasts until stable cell lines grew out.
Identification of embryonic stem cells
Pluripotency of established NTES cell lines was determined by alkaline phosphatase (ALP) staining, immunostaining and embryoid body (EB) formation. Cultured confluent ES cells were fixed with formalin for 15 min before ALP staining as described with 100 mM Tris-Hcl (pH 9.5) plus NBT (Sigma No.N-5514) and BCIP (Sigma No.B-0274) 24, 25. Immunostaining was performed with the following monoclonal antibodies: SSEA-1 (Chemicon, No. MAB4301), SSEA-3 (Chemicon, No. MAB4303), and SSEA-4 (Chemicon, No. MAB4304), Oct-4 (Santa Cruz, CA, No. SC-8628), Nanog (Gift from Tsinghua University), and followed by FITC-conjugated secondary antibody. DNA was counterstained with Propidium Iodide (10 μg/ml) or/and 0.1 μg/ml Hoechst 3332 at room temperature for 5∼10 min; Observation was performed under live cell station (Leica Co.) or confocal microscope (ZEISS, LSM 510 META). In some cell lines, the karyotype and EB formation was examined as described 3, 26, 27.
Cell cycle analysis using flow cytometry
Confluent NT-ES cells at passage 10 were trypsinized and resuspended in cold “saline GM” 28. Fixed cells in 100% ethanol overnight or for one week and to stain for DNA, cells were incubated in PBS containing 10 μg/ml of propidium iodide and 0.3 mg/ml of RNase A. To eliminate multicell aggregates, cells were filtered through a 30 μm nylon mesh; 1×104 cells were collected with a fluorescence-activated cell sorter and were analyzed using CELL QUEST 3.1 software. The percentage of cells at each cell-cycle was determined by their DNA content.
Production of chimeric mice and germline transmission confirmation
NT-ES cells were introduced into the blastocoels of CD-1 strain blastocysts by piezo-assisted microinjection. Immediately after injection, the blastocysts were transferred into pseudo-pregnant CD-1 strain surrogate mothers. When mature, chimeric offspring showing dark or gray coat colors were selected at random and were mated with CD-1 mice.
Statistic analysis
The results were analyzed using the one-way ANOVA by SPSS software.
Results
Developmental competence of reconstructed NT embryos derived from R1 ES cell donors at different passages
We examined early developmental capacity of reconstructed embryos derived from donors of relatively medium- to late-passage (15∼27) R1-ES cells, and early-passage (<5) ES cells generated from these reconstructed embryos (NT-R1-ES). We also studied reconstructed embryos derived from somatic donor cells (fresh cumulus) as well as ES donor cells generated from these somatic donor derived embryos (NTc-ES). The donor stem cell lines used for nuclear transfer were already confirmed by chimeric and germline transmission. In vivo developed embryos of the same developmental stages were used as controls.
As shown in Table 1, whereas the rate of activation and the percentage of embryos that developed to the 2-cell stage were similar for reconstructed embryos from different donor cell origins (except for those from the NT-R1-ES cells which have a lower 2-cell development rate), the developmental competencies of these different reconstructed embryos to the morula/blastocyst stages were different. In particular, the rates of morula/blastocyst formation from reconstructed embryos of somatic cells and their subsequent ES cells (NTc-ES) at medium-passage (15) were similar. Although slightly lower, this development rate was also not significantly different when using R1 ES cells at medium passages (18∼20) as NT donors. However, the rate of morula/blastocyst formation was significantly decreased for reconstructed embryos derived from R1 ES cells at late passages (25∼27). Interestingly, morula/blastocyst development seems to be improved after subsequent cloning using early passage ES cells derived from these reconstructed embryos of late-passage R1 donor, although the rates are still lower than those of the embryos derived from routine somatic stem cells or their subsequent ES cells (NTc-ES), or from medium passage R1 ES cells.
Sixteen full-term pups were obtained after nuclear transfer using medium passage (18∼20) R1-ES cells as the donor, consisting of 6.6% of the total transfer, among which, 14 survived after one week. However, after multiple attempts with several lines, no live born pups were generated using donor R1-ES cells from late passages (25∼27) or their subsequent early passage (3∼5) ES cells (NT-R1-ES) (Table 2).
Derivation of ES cell lines
Forty ES cell lines were generated, including 28 from different donor cells of the same genetic background (from in vivo developed blastocysts, SCNT blastocysts and the sequential nuclear transfer blastocysts), and 12 NT-R1-ES cell lines (Table 3). The outgrowths of fertilized blastocysts were usually cultured for 4∼5 d, and for the cloned blastocysts it takes 7∼10 d. The rates of outgrowth development were different (11.6∼81.5%) for cell lines from different donor cells, but the rates of derivation from different outgrowth were similar (42.9∼61.9%).
ESC marker expression and differentiation ability of ES cell lines
Mouse ES cells are known to express Oct4, Nanog, SSEA-1 and ALP, but not SSEA-3 and SSEA-4 29, 30. Immunostaining of these positive and negative ESC markers and ALP staining showed expected expression patterns as shown in Figures 1, 2, 3, 4 (also summarized in Table 4). In addition, the differentiation ability in terms of forming embryoid bodies (EB) when cultured in suspension was examined and all appeared positive for these cell lines (Table 4).

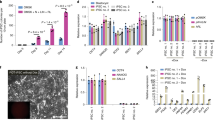
Expression of Oct4 in various mouse ES cell lines. Immunostaining with Oct4 antibody was performed on ES cells derived from fertilized embryos (A, IVP-ESCs) or cloned embryos (B, NT-ESCs; C, NT-NT-ESCs). Immunostaining with secondary antibody only is shown in D (Negative control). DNA was counterstained with Propidium Iodide (Red). Scale bars =50 μm.

Expression of SSEA1 in various mouse ES cell lines. Immunostaining with SSEA1 antibody was performed on ES cells derived from fertilized embryos (A, IVP-ESCs) or cloned embryos (B, NT-ESCs; C, NT-NT-ESCs). Immunostaining with secondary antibody only is shown in D (Negative control). DNA was counterstained with Propidium Iodide (Red). Scale bars =50 μm.

Expression of SSEA4 in various mouse ES cell lines. Immunostaining with SSEA4 a antibody was performed on ES cells derived from fertilized embryos (A, IVP-ESCs) or cloned embryos (B, NT-ESCs; C, NT-NT-ESCs). Immunostaining with secondary antibody only is shown in D (Negative control). DNA was counterstained with Propidium Iodide (Red). Scale bars =50 μm.

ALP staining of various ES cell lines. Alkaline phosphatase (ALP) staining was performed on pluripotent ES cell lines derived from fertilized embryos (A, IVP-ESCs), cloned embryos (B, NT-ESCs; C, NT-NT-ESCs,). IVP-ESCs and NT-ESCs were positive for ALP staining, while differentiated ESC were not (D, black arrows).
Karyotype analysis of ES cell lines
One of the properties of ES cells is the ability to exhibit and maintain a stable, diploid and normal complement of chromosomes (karyotype). Karyotype analysis was thus performed for the different ES cell lines generated (Table 5) and percentages of normal karyotype in our ES cell lines appear to be similar to those reported previously 3.
Cell cycle profiles of different ES cell lines
ES cells are thought to be very active in DNA synthesis and thus have a relatively long S phase, as compared with differentiated somatic cells the majority of which stay in the G0/G1 phase of the cell cycle 31. As an indicator for cell function, cell cycle analysis was performed for the various ES cell lines generated in our study. Cumulus cells of somatic origin were used as a control. As expected, the majority of the ES cells examined were in the S-phase (60%), whereas the majority of the cumulus cells were in the G0/G1 phase as reported previously (Table 6) 31 .
Discussion
NT has routinely been used to generate cloned animals, and recently with the development of therapeutic cloning, customized embryonic stem cells are produced and used for cell therapy. This provides tremendous potential for the study of mechanisms of reprogramming and development, as well as for future therapeutic efforts in regenerative medicine. However, cloning efficiency remains low, and there are also potential safety issues associated with therapeutic cloning due to known and unknown pathophysiological dysfunctions of cloned animals.
One important mean to study the molecular and cellular mechanisms and defects of reprogramming and development is through the generation and characterization of ES cell lines (NT-ESCs) by NT. NT-ESCs can be established successfully not only from cloned embryos with full developmental competency, but also from those with no potential for full-term development 3. In this study, we generated 40 ESC lines from various embryos, including those derived from R1 ES cell donors at different passages, somatic donor cells (cumulus cells) and ES cells generated from the resulting somatic-clone embryos. We examined the developmental potential of the different reconstructed embryos at several developmental stages: early zygotic gene activation (2-cell), late preimplantation (morula/blastocyst), post-implantation (implantation sites and full-term birth) and postnatal (live born, surviving pups) stages. Of the reconstructed embryos from cumulus cells, NTc-ESC, R1-ES (mid-passage at 18-20), R1-ES (late passage at 25∼27), and subsequent NT-R1-ES (early passage at 3∼5), the NT-R1-ES embryos seem to be most affected at the 2-cell stage compared to other cloned embryos (74% vs 85∼97%, respectively), which may suggest a major defect in the global reprogramming of zygotic genes in these sequentially cloned ES-derived embryos 32 (Table 1). The rate of NT embryos derived from R1 ES donors to reach the morula/blastocyst stages was significantly reduced with the late passage donors comparing with early passages. Interestingly, when used as the donor cells, the sequential early passage ESCs generated from cloned embryos (NT-R1-ESCs) have a higher rate of producing clones capable of development to blastocyst (45%) than their parental late passage R1 ES lines, indicating that a higher percentage of these sequential NT embryos overcome the 2-cell block and develop into the late preimplantation stages (Table 1). Identification of differential gene expression or gene regulation profiles in these two types of reconstructed embryos may be helpful to elucidate mechanisms governing preimplantation development. In contrast to the 6.6% of transferred reconstructed embryos that developed full term using mid-passage R1-ES donors (passage 18∼20), none of the embryos derived from the late-passage R1-ES and the subsequent NT-R1-ES donors resulted in live birth (Table 2), indicating major defects in post-implantation development in these reconstructed embryos. This result also suggests that sequential cloning using early passage ES cells (NT-R1-ES) generated from late passage ES (R1-ES)-derived reconstructed embryos fails to rescue the developmental defects that presumably originated from their parental late passage ES lines. Again, the generation of these different ES cell lines as well as their corresponding reconstructed embryos may help reveal the mechanisms governing reproduction and development of cloned embryos.
At a molecular level, various NT-ESCs derived in our study appear to have marker expression patterns resembling those ESCs that originated from in vivo developed embryos. Many of the defects demonstrated in the SCNT, ES cell or sequential ES cell NT embryos were thought to be not only due to expression of different genes, but also more importantly due to epigenetic effects. It has been reported that imprinted gene expression in ES cells is extremely unstable during their maintenance in culture 33. Epigenetic alterations in imprinted genes in ES cells persist to later developmental stages and are associated with aberrant phenotypes, since ES cells easily accumulate epigenetic alterations during in vitro culturing 34. Genomic alterations were also demonstrated in cultured human ES cells at high passages 18. Therefore, the NT-ESC lines established in our study will provide important resources for the further exploration of these factors/events at the mechanistic level.
Overall, our results showed that NT-ESCs could be established through therapeutic cloning, and for donor cells that have lost the ability to produce full term development, sequential therapeutic cloning did not appear to rescue their developmental potential.
References
Meissner A, Jaenisch R . Mammalian nuclear transfer. Dev Dyn 2006; 235:2460–2469.
Brambrink T, Hochedlinger K, Bell G, Jaenisch R . ES cells derived from cloned and fertilized blastocysts are transcriptionally and functionally indistinguishable. Proc Natl Acad Sci U S A 2006; 103:933–938.
Wakayama S, Jakt ML, Suzuki M, et al. Equivalency of nuclear transfer-derived embryonic stem cells to those derived from fertilized mouse blastocysts. Stem Cells 2006; 24:2023–2033.
Kanda S, Shiroi A, Ouji Y, et al. In vitro differentiation of hepatocyte-like cells from embryonic stem cells promoted by gene transfer of hepatocyte nuclear factor 3 beta. Hepatol Res 2003; 26: 225–231.
Daley GQ . From embryos to embryoid bodies: generating blood from embryonic stem cells. Ann N Y Acad Sci 2003; 996:122–131.
Wakayama T, Tabar V, Rodriguez I, Perry AC, Studer L, Mombaerts P . Differentiation of embryonic stem cell lines generated from adult somatic cells by nuclear transfer. Science 2001; 292:740–743.
Clark AT, Bodnar MS, Fox M, et al. Spontaneous differentiation of germ cells from human embryonic stem cells in vitro. Hum Mol Genet 2004; 13:727–739.
Toyooka Y, Tsunekawa N, Akasu R, Noce T . Embryonic stem cells can form germ cells in vitro. Proc Natl Acad Sci U S A 2003; 100:11457–11462.
Hubner K, Fuhrmann G, Christenson LK, et al. Derivation of oocytes from mouse embryonic stem cells. Science 2003; 300: 1251–1256.
Geijsen N, Horoschak M, Kim K, Gribnau J, Eggan K, Daley GQ . Derivation of embryonic germ cells and male gametes from embryonic stem cells. Nature 2004; 427:148–154.
Barberi T, Klivenyi P, Calingasan NY, et al. Neural subtype specification of fertilization and nuclear transfer embryonic stem cells and application in parkinsonian mice. Nat Biotechnol 2003; 21:1200–1207.
Odorico JS, Pirsch JD, Becker YT, et al. Results of solitary pancreas transplantation with enteric drainage: is there a benefit from monitoring urinary amylase levels? Transplant Proc 2001; 33:1700.
Takagi Y, Takahashi J, Saiki H, et al. Dopaminergic neurons generated from monkey embryonic stem cells function in a Parkinson primate model. J Clin Invest 2005; 115: 102–109.
Rideout WM, Hochedlinger K, Kyba M, Daley GQ, Jaenisch R . Correction of a genetic defect by nuclear transplantation and combined cell and gene therapy. Cell 2002; 109:17–27.
Hiendleder S, Bebbere D, Zakhartchenko V, et al. Maternal-fetal transplacental leakage of mitochondrial DNA in bovine nuclear transfer pregnancies: potential implications for offspring and recipients. Cloning Stem Cells 2004; 6:150–156.
Jouneau A, Zhou Q, Camus A, et al. Developmental abnormalities of NT mouse embryos appear early after implantation. Development 2006; 133:1597–1607.
Rehen SK, McConnell MJ, Kaushal D, Kingsbury MA, Yang AH, Chun J . Chromosomal variation in neurons of the developing and adult mammalian nervous system. Proc Natl Acad Sci U S A 2001; 98:13361–13366.
Maitra A, Arking DE, Shivapurkar N, et al. Genomic alterations in cultured human embryonic stem cells. Nat Genet 2005; 37: 1099–1103.
Niwa H, Toyooka Y, Shimosato D, et al. Interaction between Oct3/4 and Cdx2 determines trophectoderm differentiation. Cell 2005; 123:917–929.
Zhou Q, Jouneau A, Brochard V, Adenot P, Renard JP . Developmental potential of mouse embryos reconstructed from metaphase embryonic stem cell nuclei. Biol Reprod 2001; 65:412–419.
Zhou Q, Renard JP, Le Friec G, et al. Generation of fertile cloned rats by regulating oocyte activation. Science 2003; 302:1179.
Brook FA, Gardner RL . The origin and efficient derivation of embryonic stem cells in the mouse. Proc Natl Acad Sci U S A 1997; 94:5709–5712.
Wakayama T . Cloned mice and embryonic stem cell lines generated from adult somatic cells by nuclear transfer. Oncol Res 2003; 13:309–314.
Beaujean N, Bouniol-Baly C, Monod C, et al. Induction of early transcription in one-cell mouse embryos by microinjection of the nonhistone chromosomal protein HMG-I. Dev Biol 2000; 221: 337–354.
Amirand C, Viari A, Ballini JP, et al. Three distinct sub-nuclear populations of HMG-I protein of different properties revealed by co-localization image analysis. J Cell Sci 1998; 111: 3551–3561.
Dang SM, Gerecht-Nir S, Chen J, Itskovitz-Eldor J, Zandstra PW . Controlled, scalable embryonic stem cell differentiation culture. Stem Cells 2004; 22:275–282.
Magyar JP, Nemir M, Ehler E, Suter N, Perriard JC, Eppenberger HM . Mass production of embryoid bodies in microbeads. Ann N Y Acad Sci 2001; 944:135–143.
Enright BP, Jeong BS, Yang X, Tian XC . Epigenetic characteristics of bovine donor cells for nuclear transfer: levels of histone acetylation. Biol Reprod 2003; 69:1525–1530.
Thomson JA, Itskovitz-Eldor J, Shapiro SS, et al. Embryonic stem cell lines derived from human blastocysts. Science 1998; 282:1145–1147.
Munsie MJ, Michalska AE, O'Brien CM, Trounson AO, Pera MF, Mountford PS . Isolation of pluripotent embryonic stem cells from reprogrammed adult mouse somatic cell nuclei. Curr Biol 2000; 10:989–992.
Schuetz AW, Whittingham DG, Snowden R . Alterations in the cell cycle of mouse cumulus granulosa cells during expansion and mucification in vivo and in vitro. Reprod Fertil Dev 1996; 8:935–943.
Zeng F, Baldwin DA, Schultz RM . Transcript profiling during preimplantation mouse development. Dev Biol 2004; 272:483–496.
Humpherys D, Eggan K, Akutsu H, et al. Epigenetic instability in ES cells and cloned mice. Science 2001; 293:95–97.
Dean W, Bowden L, Aitchison A, et al. Altered imprinted gene methylation and expression in completely ES cell-derived mouse fetuses: association with aberrant phenotypes. Development 1998; 125:2273–2282.
Acknowledgements
This work is financially supported by grants from National Basic Research Program of China (2006CB701501) and the National Natural Science Foundation of China (30300176 and 30525040).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Zhao, C., Yao, R., Hao, J. et al. Establishment of customized mouse stem cell lines by sequential nuclear transfer. Cell Res 17, 80–87 (2007). https://doi.org/10.1038/sj.cr.7310139
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cr.7310139
Keywords
This article is cited by
-
Generation of fertile offspring from Kitw/Kitwv mice through differentiation of gene corrected nuclear transfer embryonic stem cells
Cell Research (2015)
-
Epigenetic reprogramming: roads to pluripotency
Frontiers in Biology (2010)
-
Cloning efficiency following ES cell nuclear transfer is influenced by the methylation state of the donor nucleus altered by mutation of DNA methyltransferase 3a and 3b
Frontiers in Biology (2010)
