
ABSTRACT
Genotoxic agents or inflammatory cytokines activate cellular stress responses and trigger programmed cell death. We have identified a signal transduction module, including three nuclear proteins that participate in the regulation of cell death induced by chemotherapeutic agents and tumor necrosis factor (TNF). In this nuclear signaling module, retinoblastoma protein (Rb) functions as an inhibitor of apoptotic signal transduction. Inactivation of Rb by phosphorylation or caspase-dependent cleavage/degradation is required for cell death to occur. Rb inhibits the Abl tyrosine kinase. Thus, Rb inactivation is a pre-requisite for Abl activation by DNA damage or TNF. Activation of nuclear Abl and its downstream effector p73 induces mitochondriadependent cell death. The involvement of these nuclear signal transducers in TNF induced apoptosis, which does not require new gene expression, indicates that nuclear events other than transcription can contribute to extrinsic apoptotic signal transduction.
Similar content being viewed by others

EXTRINSIC AND INTRINSIC APOPTOTIC SIGNAL TRANSDUCTION
Programmed cell death can be triggered in mammalian cells by a variety of agents. Generally speaking, the various apoptotic stimuli can be grouped into two categories: either extrinsic or intrinsic.
The cell extrinsic death signals are detected by cell surface receptors, which activate cytoplasmic signaling pathways to induce apoptosis. The paradigm for extrinsic apoptotic signal transduction is that of the FAS ligand and receptor 1. Upon binding its ligand, the FAS receptor activates “initiator” caspases (caspase-8 and -10), which transduce the death signal by cleaving other cellular proteins. FAS-mediated cell death is rapid and occurs without the requirement for new gene expression. A number of FAS related death receptors have been identified in the mammalian genomes. It is generally accepted that these receptors transduce cell extrinsic death signals through pathways similar to those of the FAS receptor.
The cell intrinsic death signals are varied and mostly generated by metabolic or genotoxic stress. The stress signals are detected and transduced by many different pathways; among them the best understood is the p53 pathway activated by genotoxic stress to induce apoptosis. There is general agreement that DNA damage activates the transcription function of p53. Among p53-regulated genes are Puma and Noxa, which encode BH3-only proteins of the Bcl2 family to induce cell death. In C. elegans, a BH3-only protein Egl1 is transcriptionally upregulated in specific cell types during development to trigger death. The intrinsic death mediated by p53-dependent upregulation of mammalian Puma or Noxa has therefore a precedent in developmentally regulated apoptosis.
In mammalian cells, disruption of mitochondria integrity is a critical event in programmed cell killing 2. Several pro-apoptotic proteins, e.g., cytochrome c, Smac, Omi, AIF and others are found in the mitochondria. When released into the cytoplasm, cytochrome c, Smac and Omi can promote the activation of caspases through the apoptosome. AIF, on the other hand, can be imported into the nucleus where it causes chromatin condensation and fragmentation. Disruption of the mitochondria membrane potential also interferes with ATP production and can further contribute to the demise of a cell. Proteins of the Bcl2-family can protect or disrupt the outer membrane of mitochondria, hence, they play critical roles in the regulation of programmed cell death. As mentioned above, BH3-only proteins of the Bcl2-family can disrupt mitochondrial integrity when they are upregulated by p53 in DNA damage-induced cell death. Cell extrinsic death signals can also disrupt mitochondria, best illustrated by the cleavage of Bid, another BH3-only protein, by activated caspase-8.
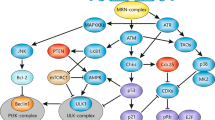
Since mitochondria disruption can contribute to death induced by either extrinsic or intrinsic signals, it is plausible that extrinsic and intrinsic death signals may be transduced to mitochondria by a common pathway. Our studies have indeed identified a signaling module that regulates apoptosis induced by tumor necrosis factor (extrinsic) and DNA damage (intrinsic). In this signaling module, the retinoblastoma protein (RB) functions as an inhibitor of apoptotic signal transduction such that RB inactivation is required for cell death. Downstream of RB, we have identified at least two proteins that activate the cell death response and they are the Abl tyrosine kinase and the p73 protein, a member of the p53-family of transcription factors. This signal transduction module participates in intrinsic as well as extrinsic apoptotic signal transduction (Fig. 1).

Nucleo-cytoplasmic communication in apoptotic signal transduction. In mammalian cells, our studies have identified a signaling module that regulates apoptosis induced by tumor necrosis factor (extrinsic) and DNA damage (intrinsic). In this signaling module, the retinoblastoma protein (RB) functions as an inhibitor of apoptotic signal transduction such that RB inactivation is required for cell death. Downstream of RB, we have identified at least two proteins that activate the cell death response and they are the Abl tyrosine kinase and the p73 protein, a member of the p53-family of transcription factors. Abl and p73 both undergo nucleo-cytoplasmic shutting. TNFR: tumor necrosis factor receptor. NES: nuclear export signal.
RB INHIBITS APOPTOTIC RESPONSE TO DNA DAMAGE AND TNF
The retinoblastoma tumor suppressor RB regulates cell proliferation and cell death 3. Bialleleic mutation of the human Rb gene causes the development of retinoblastoma in children. Inactivation of RB protein by phosphorylation is observed at G1/S transition in every cell cycle. The ability of RB to inhibit cell cycle progression underscores its tumor suppression function.
Interestingly, genetic ablation of the Rb gene in mice causes embryonic lethality that is associated with ectopic S-phase and excessive apoptosis 3. Moreover, Rb-deficient human cancer cells as well as fibroblasts derived from the Rb-knockout mouse embryos are hypersensitive to chemotherapeutic drugs. These observations suggest RB to play a role in protecting cells from apoptosis. Because proliferating cells are more sensitive to apoptosis induced by genotoxic stress, protection from DNA damage-induced cell death by RB can be explained by the anti-proliferative effect of RB. Consistent with this notion, we have found that Rb+/+ mouse embryo fibroblasts (MEFs) undergo long-term (> 6 d) arrest upon exposure to a high dose of cisplatin, etoposide or mitomycin c, whereas MEFs from their Rb−/− littermates were unable to establish long-term cell cycle arrest and continued to die during the experimental time course 4. Thus, RB protects cells against genotoxin-induced death most likely because it can inhibit cell cycle progression.
Independent of cell cycle arrest, RB can also directly inhibit apoptotic signal transduction as demonstrated with TNF-induced cell death. This study began with the observation that activation of caspase leads to RB cleavage and degradation 5. Several caspase cleavage sites have been mapped in the RB protein. A DEADG motif at the extreme C-terminus of RB was found to be the predominant cleavage site when cells are activated by extrinsic death signals, such as TNF, that rely on caspases for the transduction of death signal. Substitution mutation at this site generated the RB-MI protein (DEAAE) that is resistant to caspase cleavage 6. Ectopic expression of a cDNA encoding RB-MI protected fibroblasts from TNF-induced apoptosis 6. RB-MI also protected granular neurons from depolarization-induced apoptosis in primary culture 7.
The MI-substitution mutation has been introduced into the mouse Rb gene through Escell based knock-in technology 8. The Rb-MI mice developed normally without any obvious defects. When challenged with lipopolysaccharide (LPS), an inducer of inflammation and septic shock, male Rb-MI mice showed increased survival relative to their wild type littermates. The physiological basis for this male-specific resistance of Rb-MI mice to septic shock is presently not understood. When tissue apoptotic response was examined, the intestinal tracts of LPS-treated male and female Rb-MI mice showed reduced levels of TUNEL-positive cells. Intestinal apoptosis in LPS-treated mice is dependent on TNF and type I TNF-receptor (TNFRI). The reduced intestinal apoptosis in LPS-treated Rb-MI mice therefore provides in vivo evidence that RB-MI can protect cells from TNF-induced death.
Fibroblasts derived from the Rb-MI embryos (MEFs) were found to be also resistant to TNF-induced apoptosis 8. In MEFs, the Rb-MI protein is resistant to degradation when TNFRI is stimulated either by binding to human TNF or to agonistic TNFRI antibody 8. The Rb-MI protein is still degraded when TNFRI and TFNRII are both engaged either by binding to murine TNF or to agonistic antibodies against both types of TNF receptors 8. As a result, Rb-MI MEFs are protected from apoptosis induced by human TNF but not murine TNF 8. Clearly, Rb-MI is not completely resistant to degradation. However, when Rb-MI is not degraded, TNF-induced apoptosis is prevented. When Rb-MI is degraded, TNF-induced apoptosis is restored.
Previous studies have shown that RB is also cleaved by caspase in response to genotoxic stress 5. We have found that Rb-MI is resistant to cleavage in MEFs treated with doxorubicin, which is an inhibitor of topoisomerase II that triggers DNA damage-induced apoptotic program. Despite the preservation of Rb-MI, we did not observe any reduction in doxorubicin-induced apoptosis 8. In mice, the neonatal retinal blasts readily undergo apoptosis in response to ionizing radiation 9. Again, this response is not abrogated in the Rb-MI mice 8. We have found that Fas-induced apoptosis in hepatocyotes was not reduced in Rb-MI mice. Moreover, TRAIL-induced apoptosis in MEFs also proceeded without impediments despite the expression of Rb-MI. Taken together, these results suggest Rb-MI specifically interferes with the transduction of death signal from the type I TNF receptor. We have found two pro-apoptotic signal transducers, Abl and p73, that are negatively regulated by RB. Interestingly, Abl and p73 both contribute to DNA damage and TNF-induced apoptosis.
DNA DAMAGE AND TNF INDUCED APOPTOSIS INVOLVES ABL
The ubiquitously expressed Abl tyrosine kinase is a transducer of a variety of cell extrinsic and intrinsic signals including those from growth factors, cell adhesion, oxidative stress and DNA damage 10, 11. The Abl protein contains three nuclear localization signals (NLS), one nuclear export signal (NES) and undergoes nucleocytoplasmic shuttling 12. The N-terminal region of Abl resembles the Src-family of tyrosine kinases and adopts an auto-inhibited conformation assembled through intramolecular interactions among its SH3-SH2-kinase domains 13, 14. The C-terminal region of Abl contains subcellular location cues, including NLS, NES, binding sites for G-actin, F-actin and binding site for DNA 10, 11. We have shown that F-actin, but not G-actin, is an allosteric inhibitor of Abl tyrosine kinase 15. F-actin functions as a “coinhibitor”, enforcing or stabilizing the “auto-inhibited” conformation to inhibit Abl kinase activity 14. The cytoplasmic Abl tyrosine kinase regulates F-actin dynamics in response to growth factors and cell adhesion 10.
In the nucleus, Abl interacts with RB, which binds to the Abl kinase domain and inhibits its activity 16. The nuclear Abl kinase is activated by DNA damage 17. Activation of nuclear Abl kinase by ionizing radiation (IR) is dependent on a functional ATM, which phosphorylates Abl at a serine site (Ser465) in the kinase domain 18. Substitution of Ser465 with Ala or Glu abolished DNA damage-induced activation of Abl kinase; moreover, Glu substitution was able to increase the basal activity of Abl. While Abl is dispensable for IR-induced apoptosis in neonatal retinas of the mice 9, activation of Abl contributes to the apoptosis response to several chemotherapeutic agents including cisplatin, cytosine arabinose, camptothecin and doxorubicin in cultured cells 19, 20. The direct binding and inhibition of Abl kinase may therefore account in part for the antiapoptotic function of RB.
The nuclear Abl tyrosine kinase is also activated by TNF 21, 22. Inhibition of Abl kinase by imatinib, a small molecular inhibitor, can attenuate the death response to TNF in human myeloid leukemia U937 cells and Rb-knockout MEFs 21, 22. MEFs derived from Rb−/−Abl−/−embryos are resistant to TNF-induced apoptosis while MEFs from Rb−/−Abl+/+ littermate embryo are sensitive to TNF 22. Moreover, thymocytes from Abl−/−mice displayed a reduced death response to TNF when compared to thymocytes from littermate Abl+/+ mice 22. The requirement for Abl in TNF-induced death is not universal because we have found that imatinib does not inhibit the apoptotic response of 3T3 cells (immortalized MEFs) to TNF plus cycloheximide (CHX). Moreover, siRNA knockdown of Abl does not interfere with TNF/CHX-induced death in HCT116 colon carcinoma cells. At present, we do not understand the differential requirement of Abl for TNF-induced apoptosis. However, our observation is in keeping with other reports that suggest TNF may activate several parallel mechanisms to kill cells. Our observations suggest that at least one but not all of TNF-activated death mechanisms are dependent on Abl.
As discussed above, proteolytic cleavage and degradation of Rb is required for TNF to induce apoptosis. We have found in Rb-MI MEFs, nuclear Abl tyrosine kinase is not activated by TNF 22. This suggests that Rb cleavage/degradation is a pre-requisite for TNF to activate Abl kinase. With Rb-knockout MEFs, we observed a higher basal activity of nuclear Abl kinase that was still activated by TNF 22. This suggests that the pro-apoptotic function of nuclear Abl kinase to be controlled by a hierarchy of negative and positive regulators. The negative regulation of Abl by Rb must be neutralized through Rb cleavage/degradation before the positive regulation of Abl by TNF can be achieved (Fig. 1). At present, we do not know the identity of the positive regulator that mediates the activation of nuclear Abl kinase following Rb degradation. As discussed above, the positive regulator of nuclear Abl kinase in DNA damage response is the ATM kinase. Whether ATM or a related activity mediates the activation of nuclear Abl by TNF remains to be determined.
DNA DAMAGE AND TNF INDUCED APOPTOSIS INVOLVES p73
We have identified a third protein—p73 that regulates the apoptotic response to DNA damage and TNF. The p73 protein belongs to the p53-family of transcription factors 23. Exposure to chemotherapeutic agents such as cisplatin, camptothecin, and doxorubicin, causes the stabilization and activation of the p73 protein, a process that requires Abl 19, 20, 24. Ectopic expression of p73 leads to variable levels of cell death in different cell lines. The apoptotic function of ectopically expressed p73 can be enhanced significantly by the co-expression of Abl tyrosine kinase or by exposure to DNA damaging agents. MEFs derived from p73-knockout mice are capable of stabilizing and activating p53 to undergo apoptosis when exposed to doxorubicin. However, reintroduction of p73 caused a much greater death response to the drug and this sensitization could be blocked by the inhibition of Abl kinase. The cumulated evidence supports interdependence between Abl and p73 in activating apoptosis following DNA damage.
Three lines of evidence have established a requirement of p73 for TNF-induced apoptosis. The apoptotic response of p73-knockout MEFs to TNF/CHX is enhanced upon the reintroduction of p73 expression 22. Knockdown of p73 by siRNA reduced the apoptotic response of human cancer cells HCT116 to TNF/CHX 22. Thymocytes derived from p73-knockout mice are resistant to apoptosis induced by TNF 22. While p73-knockout thymocytes are protected from TNF, they display wild type level of apoptosis in response to ionizing radiation (IR) 25. By contrast, p53 is required for IR to induce thymocyte apoptosis, but p53-knockout thymocytes are sensitive to TNF induced cell death 22. It thus appears that p73 and p53 have non-redundant functions in the transduction of death signals in thymocytes: p73 transduces death signal from TNF but not from IR whereas p53 transduces death signal from IR but not from TNF. This rule may not apply to all cell types, because p73 has been implicated in IR-induced apoptosis in MCF7 human breast cancer cells 26.
The mechanism by which p73 activates apoptosis is not completely understood. Several studies have demonstrated that p73 can activate transcription from p53-regulated promoters, e.g., p21Cip1, p53AIP, Bax, Puma, etc 23. Recent studies have demonstrated that Puma is required for IR-induced apoptosis 27. As mentioned above, IR-induced apoptosis in MCF7 cells has been shown to require p73 26. Therefore, transactivation of pro-apoptotic gene expression by p73 may account for its pro-apoptotic function. This, however, does not readily explain how p73 contributes to TNF-induced apoptosis. Extrinsic death signal transduction occurs in the absence of new protein synthesis. As discussed above, cycloheximide enhances rather than prevents TNF-induced apoptosis. The requirement for p73 in TNF-induced apoptosis indicates that p73 may alos promote death signal transduction through transcription-independent mechanisms.
NUCLEAR REGULATION OF MITOCHONDRIA INTEGRITY
Our finding suggests that a nuclear signaling module, including Rb, Abl and p73, participate in the transduction of TNF and DNA damage signals to disrupt the mitochondria. TNF-induced cytochrome c release and pro-caspase 3 cleavage are compromised in cells that cannot degrade Rb (the Rb-MI cells), cannot activate Abl (Ablknockout cells), or lack p73 (p73-knockout cells) 8, 22. Therefore, degradation of nuclear Rb, and activation of nuclear Abl and p73 can lead to the disruption of mitochondria in the cytoplasm. The most obvious involvement of nuclear events in mitochondrial regulation is the trans-activation of BH3-only proteins such as Puma, which can stimulate cytochrome c release. However, because TNF-induced cell death can proceed without new gene expression, the nuclear signaling module may regulate mitochondria integrity by additional mechanisms.
As a working hypothesis, we propose that certain regulators of mitochondrial integrity may be sequestered in the nucleus. This would be analogous to the sequestration of transcription factors in the cytoplasm, albeit the roles of nucleus and the cytoplasm would be reversed. In as much as extracellular signals can promote the nuclear translocation of transcription factors to activate gene expression, nuclearly sequestered “mitochondria wracking factors (MWFs)” may be released into the cytoplasm in response to death signals. If so, the release of one or more of these MWFs might be regulated by the degradation of Rb and the activation of nuclear Abl and p73.
The Abl tyrosine kinase and the p73 protein both contain nuclear localization and nuclear export signals and undergo nucleo-cytoplasmic shuttling 12, 28 (Fig. 1). Recently, several reports have suggested that p53 can directly participate in the disruption of mitochondrial outer membranes by acting in the cytoplasm 29. We did not observe a significant exit of p73 from the nucleus in response to TNF/CHX (B. Nelson Chau & Jean Y.J.Wang, unpublished). However, this does not rule out the possibility that a small fraction of p73 is released into the cytoplasm to disrupt mitochondrial integrity in response to death signals. In ongoing studies, we have observed that Abl tyrosine kinase must be activated in the nucleus to induce cell death. In other words, the nuclear localization of Abl is critical to its death inducing function. Interestingly, the nuclear export of Abl also contributes to apoptosis, suggesting that exit of activated Abl from the nucleus contributes to apoptotic signal transduction. Whether Abl and p73 are themselves MWFs is an interesting question that is being addressed.
References
Hengartner MO . The biochemistry of apoptosis. Nature 2000; 407:770–6.
Wang X . The expanding role of mitochondria in apoptosis. Genes Dev 2001; 15:2922–33.
Chau BN, Wang JY . Coordinated regulation of life and death by RB. Nat Rev Cancer 2003; 3:130–8.
Knudsen KE, Booth D, Naderi S, et al. RB-dependent S-phase response to DNA damage. Mol Cell Biol 2000; 20:7751–63.
Tan X, Wang JY . The caspase-RB connection in cell death. Trends Cell Biol 1998; 8:116–20.
Tan X, Martin SJ, Green DR, Wang JY . Degradation of retinoblastoma protein in tumor necrosis factor- and CD95-induced cell death. J Biol Chem 1997; 272:9613–6.
Boutillier AL, Trinh E, Loeffler JP . Caspase-dependent cleavage of the retinoblastoma protein is an early step in neuronal apoptosis. Oncogene 2000; 19:2171–8.
Chau BN, Borges HL, Chen TT, et al. Signal-dependent protection from apoptosis in mice expressing caspase-resistant Rb. Nat Cell Biol 2002; 4:757–65.
Borges HL, Chao C, Xu Y, Linden R, Wang JY . Radiation-induced apoptosis in developing mouse retinaexhibits dose-dependent requirement for ATM phosphorylation of p53. Cell Death Differ 2004; 11:494–502.
Woodring PJ, Hunter T, Wang JY . Regulation of F-actin-dependent processes by the Abl family of tyrosine kinases. J Cell Sci 2003; 116:2613–26.
Zhu J, Wang JY . Death by Abl: a matter of location. Curr Top Dev Biol 2004; 59:165–92.
Taagepera S, McDonald D, Loeb JE, et al. Nuclear-cytoplasmic shuttling of C-Abl tyrosine kinase. Proc Natl Acad Sci U S A 1998; 95:7457–62.
Nagar B, Hantschel O, Young MA, et al. Structural basis for the autoinhibition of c-Abl tyrosine kinase. Cell 2003; 112:859–71.
Wang JY . Controlling Abl: auto-inhibition and co-inhibition? Nat Cell Biol 2004; 6:3–7.
Woodring PJ, Hunter T, Wang JY . Inhibition of c-Abl tyrosine kinase activity by filamentous actin. J Biol Chem 2001; 276:27104–10.
Welch PJ, Wang JY . A C-terminal protein-binding domain in the retinoblastoma protein regulates nuclear c-Abl tyrosine kinase in the cell cycle. Cell 1993; 75:779–90.
Wang JY . Regulation of cell death by the Abl tyrosine kinase. Oncogene 2000; 19:5643–50.
Baskaran R, Wood LD, Whitaker LL, et al. Ataxia telangiectasia mutant protein activates c-Abl tyrosine kinase in response to ionizing radiation. Nature 1997; 387:516–9.
Gong JG, Costanzo A, Yang HQ, et al. The tyrosine kinase c-Abl regulates p73 in apoptotic response to cisplatin-induced DNA damage. Nature 1999; 399:806–9.
Truong T, Sun G, Doorly M, Wang JY, Schwartz MA . Modulation of DNA damage-induced apoptosis by cell adhesion is independently mediated by p53 and c-Abl. Proc Natl Acad Sci U S A 2003; 100:10281–6.
Dan S, Naito M, Seimiya H, Kizaki A, Mashima T, Tsuruo T . Activation of c-Abl tyrosine kinase requires caspase activation and is not involved in JNK/SAPK activation during apoptosis of human monocytic leukemia U937 cells. Oncogene 1999; 18:1277–83.
Chau BN, Chen TT, Wan YY, DeGregori J, Wang JY . Tumor necrosis factor alpha-induced apoptosis requires p73 and c-Abl activation downstream of RB degradation. Mol Cell Biol 2004; 24:4438–47.
Melino G, De Laurenzi V, Vousden KH . p73: Friend or foe in tumorigenesis. Nat Rev Cancer 2002; 2:605–15.
Vella V, Zhu J, Frasca F, et al. Exclusion of c-Abl from the nucleus restrains the p73 tumor suppression function. J Biol Chem 2003; 278:25151–7.
Senoo M, Manis JP, Alt FW, McKeon F . p63 and p73 are not required for the development and p53-dependent apoptosis of T cells. Cancer Cell 2004; 6:85–9.
Agami R, Blandino G, Oren M, Shaul Y . Interaction of c-Abl and p73alpha and their collaboration to induce apoptosis. Nature 1999; 399:809–13.
Jeffers JR, Parganas E, Lee Y, et al. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell 2003; 4:321–8.
Inoue T, Stuart J, Leno R, Maki CG . Nuclear import and export signals in control of the p53-related protein p73. J Biol Chem 2002; 277:15053–60.
Chipuk JE, Green DR . Cytoplasmic p53: bax and forward. Cell Cycle 2004; 3:429–31.
Acknowledgements
Our work on the regulation of apoptosis by Rb, Abl and p73 is supported by grants from the National Cancer Institute, USA (CA43054 and CA58320). Dr. B. Nelson Chau, a postdoctoral fellow of the Damon Runyon Research Foundation, has made important contributions to this study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
WANG, J. Nucleo-cytoplasmic communication in apoptotic response to genotoxic and inflammatory stress. Cell Res 15, 43–48 (2005). https://doi.org/10.1038/sj.cr.7290263
Issue Date:
DOI: https://doi.org/10.1038/sj.cr.7290263
Keywords
This article is cited by
-
The efficacy of Epigallocatechin-3-gallate (green tea) in the treatment of Alzheimer’s disease: an overview of pre-clinical studies and translational perspectives in clinical practice
Infectious Agents and Cancer (2017)
-
Genetic disruption of Abl nuclear import reduces renal apoptosis in a mouse model of cisplatin-induced nephrotoxicity
Cell Death & Differentiation (2013)
-
Selective Targeting of c-Abl via a Cryptic Mitochondrial Targeting Signal Activated by Cellular Redox Status in Leukemic and Breast Cancer Cells
Pharmaceutical Research (2012)
-
An evolutionarily acquired genotoxic response discriminates MyoD from Myf5, and differentially regulates hypaxial and epaxial myogenesis
EMBO reports (2011)
-
Eph tumour suppression: the dark side of Gleevec
Nature Cell Biology (2006)
