
Abstract
Major histocompatibility complex class II locus DRB variation was investigated by single-strand conformation polymorphism analysis and sequence analysis in the two subspecies of Pyrenean chamois (Rupicapra pyrenaica) endemic to the Iberian Peninsula. Low levels of genetic variation were detected in both subspecies, with seven different alleles in R. p. pyrenaica and only three in the R. p. parva. After applying the rarefaction method to cope with the differences in sample size, the low allele number of parva was highlighted. The low allelic repertoire of the R. p. parva subspecies is most likely the result of bottlenecks caused by hunting pressure and recent parasitic infections by sarcoptic mange. A phylogenetic analysis of both Pyrenean chamois and DRB alleles from 10 different caprinid species revealed that the chamois alleles form two monophyletic groups. In comparison with other Caprinae DRB sequences, the Rupicapra alleles displayed a species-specific clustering that reflects a large temporal divergence of the chamois from other caprinids, as well as a possible difference in the selective environment for these species.
Similar content being viewed by others

Introduction
The major histocompatibility complex (MHC) is the foremost genetic component of the mammalian immune system, as the genes located in this genomic region are crucial for the initiation of the adaptive immune response. In humans and in domestic ungulates, the MHC class II DRB1 homologues are the most polymorphic loci in the entire genome, and much of the variation is located in the functionally important peptide-binding regions (PBR). Most natural mammal populations studied also possess high MHC diversity, in terms of both the number of alleles present and the extent of sequence variation among alleles (Potts and Wakeland, 1990). The high polymorphism detected in the MHC region has been associated with the effectiveness of its immunological self/non-self-recognition function. It appears to change rapidly in response to evolving infectious agents that periodically affect natural populations (Yuhki and O'Brien, 1990), suggesting that MHC variability is maintained by natural selection. Given a connection between MHC variability and the ability of the immune system to respond to a higher variety of pathogens, it has been suggested that species or populations with low MHC diversity might be particularly vulnerable to infectious diseases (O'Brien and Evermann, 1988; Gutierrez-Ezpeleta et al., 2001).
However, limited MHC polymorphism in the Eurasian beaver (Ellegren et al., 1993), moose (Mikko and Andersson, 1995), musk ox, roe or fallow deer (Mikko et al., 1999) appears not to have hampered population expansion following recent bottlenecks. Data from a broader range of species living in different environments and with different lifestyles might, therefore, shed more light on the causes and consequences of reduced MHC polymorphism in natural populations. Accordingly, several studies have been performed in the last 5 years on the MHC-DRB1 gene of different wild caprinids: bighorn sheep (Ovis Canadensis; Gutierrez-Ezpeleta et al., 2001), Spanish ibex (Capra ibex; Amills et al., 2004), Soay sheep (Charbonnel and Pemberton, 2005) and Alpine chamois (Rupicapra rupicapra, Schaschl et al., 2004). A limited allelic repertoire of the DRB1 gene was detected in the Spanish ibex (six alleles, He=0.562–0.659), while extensive allelic and nucleotide variability was found in bighorn sheep (21 different alleles, He=0.742 in a sample of 213 animals) and Alpine chamois (19 alleles in 59 animals). These results were unexpected and may reflect responses to different factors such as fragmented habitats, population crashes or a possible relaxed pathogen-driven selection in Alpine habitats.
Chamois (genus Rupicapra) are mountain ungulates presently distributed over most of the medium- to high-altitude mountain ranges of Southern Europe, the Balkans and the Middle East. There are two extant chamois species: R. rupicapra (Alpine chamois) and Rupicapra pyrenaica (Pyrenean chamois) (Masini and Lovari, 1988). Although long considered to be subspecies, they are now classified as distinct species, probably separated during the Middle Pleistocene. The R. pyrenaica species consists of three subspecies (pyrenaica, parva and ornata), two of which are endemics of the Iberian Peninsula: R. p. pyrenaica in the Pyrenees and Rupicapra pyrenaica parva, in the Cantabrian mountains. The third (Rupicapra pyrenaica ornata) is restricted to the Massif of Abruzzo, Italy. The Iberian subspecies differ in morphology (the Cantabrian chamois is smaller) and in the color of the coat, being reddish in summer and light gray in winter in the Pyrenean population, and yellowish and dark gray in the Cantabrian population.
The health of the populations also differs. The population dynamics of R. p. parva, like R. rupicapra populations in large parts of the eastern Alps, are influenced by sarcoptic mange epidemics, an infection of Sarcoptes rupicaprae (Fernández-Morán et al., 1997). However, sarcoptic mange has not been detected in the Pyrenean chamois R. p. pyrenaica (Arnal and Fernández de Luco, 2004).
The analysis of genetic variability in two subspecies with different demographic histories could reveal the genetic impact of demographic bottlenecks and the evolution of MHC polymorphism.
Thus, this study has two aims:
-
1)
To estimate the MHC variability of the two R. pyrenaica subspecies of the Iberian Peninsula, and
-
2)
To analyze the evolution of DRB gene polymorphism in Pyrenean chamois, in comparison with the Alpine chamois.
Materials and methods
Samples and collection sites
The total population of chamois in the Pyrenees in Aragon (Spain) is estimated to be nearly 15 000 animals. A number of Natural Management Areas (NMA) have been delineated to encompass mountain massifs, as the main annual movements are migrations in altitude (Herrero et al., 2004). Overall, around 500 animals (males and females) are hunted every year (Herrero et al., 2004). When possible, samples were collected by gamekeepers and stored in alcohol. Samples from animals included in this study all came from sport hunting practiced in game reserves managed by the regional government of Aragon and were located in nine different management areas along the French/Spanish border. Our objective was to collect at least 10 random samples from each location, but we obtained smaller samples in some of the NMA because of smaller population size (Peña Forca and Punta Suelsa) or because of national park status (Monte Perdido).
The current estimated population size of R. p. parva is about 6000 individuals (Pérez et al., 2002). The samples included in this study all came from sport hunting practiced in game reserves managed by the regional government of Asturias, and were kindly donated by Dr A Domínguez (University of Oviedo, Spain).
Muscle tissue samples were derived from 98 free-living Pyrenean chamois (R. p. pyrenaica subspecies) shot between 1997 and 2000, and from 32 free-living chamois of the R. p. parva subspecies, which were shot in 1995 in three different hunting areas of the Cantabrian mountains in eastern Asturias (see Figure 1 and Table 1).

Sampling locations, as follows: R. p. parva in the Cantabrian mountains. (A) Somiedo (1) located in the west (CBW), and Aller (2) and Caso (3) located in the east (CBE); R. p. pyrenaica (B) in the Aragonese Pyrenees: Peña Forca (4), Bixaurin (5), Anayet (6), Biñamala (7), Monte Perdido (8), Liena (9), Punta Suelsa (10), Posets (11) and Maladeta (12).
PCR, SSCP and sequencing
Genomic DNA was extracted from chamois samples using the phenol/chloroform method (Sambrook et al., 1989). After testing different sets of primers, and due to the high number of alleles amplified in sheep (Arrieta-Aguirre et al., 2006), the goat primers DRB 1.1 and 1.2 (Amills et al., 1995) were chosen for this study. PCR amplifications were conducted in 50 μl reactions containing 5 μl PCR buffer, 100 ng of genomic DNA and final concentrations of 0.12 μ M primer, 100 μ M deoxynucleotides, 1.0 mM MgCl2 and 1.5 units of Taq DNA polymerase (Bioline, London, UK). After preheating at 94°C for 5 min, 40 amplification cycles were performed, followed by incubation at 72°C for 10 min. Each amplification cycle consisted of three steps: 60 s at 94°C for denaturing, 90 s at 60°C for annealing and 90 s at 72°C for elongation.
Single-strand conformation polymorphism (SSCP) analysis was carried out according to the method described by Orita et al. (1989), albeit with some modifications. A total of 1 μl of amplified solution was mixed with 6 μl of denaturing solution (USB, Cleveland, Ohio, USA) containing formamide. The mixtures were denatured at 95°C for 5 min, immediately cooled on ice and applied to a 12% nondenaturing polyacrylamide minigel. Electrophoresis was carried out for 130 min at 300 V, in 1 × Tris-Borate-EDTA buffer and at a constant room temperature of 17°C. Single-strand DNA fragments were detected by silver staining. All the animals presented two or four bands (homozygous with two single-stranded bands or heterozygous with two pairs of single-stranded bands), suggesting that we were analyzing a single locus, which was named MhcRupy-DRB1 (Klein et al., 1990). No deletions or stop codons were found among these nucleotide sequences, suggesting that we were not amplifying a pseudogene. However, no expression study has been performed, so the sequences obtained are putative MHC genes.
PCR amplicons of each SSCP pattern were cleaned using the QIAquick PCR Purification Kit (Qiagen, GmbH, Hilden, Germany). Purified PCR products were sequenced in both directions in an ABI 310 DNA Sequence Analyzer (Applied Biosystems, Foster City, CA, USA) automated sequencer, following the ABI PRISM BigDye Terminator Cycle Sequencing Ready Reaction Kit (Applied Biosystems) protocol. The sequences of the three most common alleles were obtained by analyzing independent PCR products from nine homozygous samples. Homozygous samples were directly sequenced on both strands. The sequence of the remaining five alleles, present only in heterozygous individuals, was deduced by comparing the heterozygous individuals detected by SSCP with one allele in common with a corresponding homozygous. At least two independent amplicons were analyzed for each SSCP pattern, corresponding to different individuals or different amplifications of the same individual.
Statistical and phylogenetic analysis
Different measures of genetic diversity were estimated: number of alleles by direct counting, effective number of alleles (ne), number of alleles by rarefaction, and observed and expected heterozygosity. The effective number of alleles (ne) is the number of equally frequent alleles that would be required to produce the same heterozygosity as observed in an actual population, and is calculated as the reciprocal of the expected homozygosity ne=1/∑fi2. The rarefaction method (Hurlbert, 1971; Petit et al., 1998) was employed to correct the variation in sample size in the measurement of the number of alleles or allelic richness. A simplified version of the CONTRIB program was used to make these computations and is available at www.pierroton.inra.fr/labo/software. Deviation from Hardy–Weinberg equilibrium was tested using the Markov chain permutation test in GENEPOP (Raymond and Rousset, 1995).
A population phylogenetic tree was created using the neighbor-joining method based on the Da distance (Nei et al., 1983) calculated among all sampled populations with the statistical package populations (Langella, 2002). The Da distance is a recommended distance measure for closely related or small populations. The robustness of the tree was evaluated by carrying out 1000 bootstrap iterations.
Rupy-DRB1 nucleotide sequences were analyzed and aligned in BIO EDIT version 5.0.9 (Hall, 1999) and translated into the corresponding amino acid sequences using the MEGA software package (Kumar et al., 2004). Amino acid positions involved in peptide binding were identified by comparison with the peptide-binding groove structure of humans (Brown et al., 1993). The relative frequencies of nonsynonymous (dN) and synonymous (dS) substitutions were calculated for the PBR and non-PBR for the whole exon and for the β-sheet and the α-helix segments according to Nei and Gojobori (1986) method and using the Jukes and Cantor (1969) correction; the statistical significance of observed differences in d, dN or dS was tested as described in Kumar et al. (2004).
Phylogenetic relationships between all Pyrenean chamois alleles (four from Schaschl et al., 2005; eight from this study) were reconstructed using three independent phylogenetic methods (neighbor-joining based on Kimura's two distance parameters; maximum parsimony and maximum likelihood) by means of MEGA 3.1 (Kumar et al., 2004), PAUP 4b10 (Swofford, 1998) and PHYLM 2.2.4 (Guindon and Gascuel, 2003) software programs.
Divergence times between allelic lineages of chamois were estimated using a linearized neighbor-joining tree (Takezaki et al., 1995). The rate used for dating divergence times was 0.97 × 10−9 substitutions per site per year, estimated substitution rate for DRB including PBR (Satta et al., 1991). All published sequences of bighorn sheep (O. canadensis, Ovca), domestic and wild goat (Capra hircus, Cahi; Capra aegagrus, Caae; Capra pyrenaica, Capy), musk ox (Ovibos moschatus, Ovmo), argali (Ovis ammon), mufflon (Ovis orientalis musimon), Alpine chamois (R. rupicapra, Ruru) and Pyrenean chamois (R. pyrenaica, Rupy) were included in this analysis. To give the two domestic species (sheep and goat) the same weight, the domestic sheep (Ovar) sequence number was reduced to equal the goat sequence number, and selected to reflect all the major allelic types described so far. Alleles grouped within a major type differ by less than five amino acid substitutions, and the first two digits after the asterisk specify which major allelic type the allele belongs to; for a revision table see Konnai et al. (2003). Relative support of the branching order was estimated using bootstrap analysis with 1000 iterations.
Results
Genetic variability of the Iberian Pyrenean chamois
PCR–SSCP and sequence analysis of the Rupy-DRB1 gene in the two Iberian subspecies of Pyrenean chamois allowed us to detect eight distinct alleles (GenBank accession numbers AY212149; AY212150; AY212152-AY212157). We gave each allele a name based on its sequence: Rupy-DRB1*01 to Rupy-DRB1*08. Among the most frequent alleles, alleles Rupy-DRB1*02 and Rupy-DRB1*04 were shared by both Iberian subspecies, but allele Rupy-DRB1*01, quite frequent in most of the Pyrenean populations, was absent in parva, and allele Rupy-DRB1*05 was absent in pyrenaica (see Table 1). In total, seven and three alleles were detected in the P. p. pyrenaica and P. p. parva subspecies, respectively, although ne, a measure which takes into account allele frequencies, was quite similar in both subspecies: 2.54 and 2.11, respectively. To cope with uneven sample sizes (98 and 32 animals), allelic richness was standardized by the rarefaction method. We estimated this parameter from a sample of 64 gene copies corresponding to the smallest sample size studied (the smallest group comprising 32 individuals, that is 64 gene copies). In this case, the number of alleles for R. p. pyrenaica subspecies was five, still higher than the three alleles detected in R. p. parva. Notably, only two alleles were detected in the Aller and Caso Cantabrian subpopulations.
Although observed heterozygosity (Ho) was higher than expected from Hardy–Weinberg equilibrium (He), there were few significant deviations from Hardy–Weinberg expectations, with the exception of the Aller population of R. p. parva, where there was a significant deficit of heterozygotes (P<0.01) (Table 2).
The Da genetic distance (Nei et al., 1983) between the three Cantabrian subpopulations averaged 0.1133, whereas it averaged 0.0795 between the nine Pyrenean subpopulations, so the Cantabrian subpopulations were more different from one another than the Pyrenean subpopulations. In fact, three main clusters could be observed in the dendrogram based on Da distances (Figure 2): the two eastern Cantabrian subpopulations, the western subpopulation and the Pyrenean ones. However, although all Pyrenean subpopulations cluster together, there is little geographical clustering among them.

Neighbor-joining tree based on Da genetic distance for the 12 chamois populations included in the study.
Evolution of the MhcRupy-DRB1 gene
At the sequence level, we found that 26 of the 237 nucleotidic sites (11%) analyzed were variable, and 17 sites out of 79 (21.5%) of the inferred amino acid sequences were variable. Fourteen out of seventeen variable amino acid sites (82.35%) were found within the putative PBR (see Figures 3a and 4). Although the number of alleles in the Pyrenean chamois was small, the difference between these alleles was relatively large, with a difference in amino acid sequence of 4–13 amino acids.

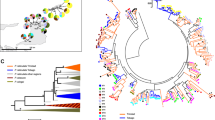
Alignment of amino acid sequences encoded by the eight DRB exon 2 alleles of Pyrenean chamois (R. pyrenaica) detected in this work. A point is used to show identity to Rupy-DRB1*01. Amino acid residues forming the putative PBR (Brown et al., 1993) are identified by a cross. Allele frequencies in each subspecies have been included (pyr, pyrenaica; par, parva). Alleles detected by Schaschl et al. (2005) have also been included for comparative purposes.

Neighbor-joining tree for the second exon of Pyrenean chamois DRB alleles. The eight alleles detected in this study and four alleles from Schaschl et al. (2005) have been included. The tree has been rooted with a bovine sequence. The topology of the tree is supported by bootstrap P-values (1000 iterations). Values below represent bootstrap values of maximum parsimony and maximum likelihood analysis, in this order.
The most frequent allele in both subspecies (Rupy-DRB*02) had two amino acid residues unique to chamois alleles in comparison with other ruminants: amino acid L in position 37 and F in position 78; for comparative tables see Jugo and Vicario (2000). Moreover, among ruminants position 41 was polymorphic only in chamois. The relative frequency of nonsynonymous substitutions for the PBR region was seven times that of synonymous substitutions (dN/dS=7.23) (see Table 3). As it has been proposed that the β-strand and the α-helical regions in the first domain of polymorphic class II β-chains have different evolutionary histories, we estimated the substitutions for these two regions independently. The number of replacement sites in the α-helix was similar to that of the β-sheet at the PBR in the two species. The number of synonymous changes within exon 2 of both Rupicapra species alleles falls far below the rate of substitutions of other ruminants. These findings suggest that the diversity of Rupicapra alleles has evolved more recently than that of other ruminant species.
Figure 4 depicts the phylogenetic relationship of all R. pyrenaica alleles with a bovine allele as an outgroup. Two main clusters could be discerned, with one of them being subdivided into several subgroups. Although some branches were not supported by the bootstrap analysis, it seemed that the Pyrenean chamois DRB1 sequences represented at least three major allelic lineages, defined as clades with >70% boostrap support: Rupy-DRB1*5 and Rupy-DRB1*7; Rupy-DRB1*08, Rupy-DRB1*11, Rupy-DRB1*04 Rupy-DRB1*06 and Rupy-DRB1*13; and Rupy-DRB1*02 and Rupy-DRB1*10. Figure 5 presents a linearized neighbor-joining tree with all known Rupicapra alleles and homologous class II DRB exon 2 sequences from other caprinids. The R. rupicapra and R. pyrenaica alleles were intermingled: all alleles formed one of two monophyletic groups, and were not scattered among alleles from other species. Interestingly, a very well supported old allelic lineage including some sheep alleles, a few goat alleles and the mufflon sequence could be identified in this tree.

Linearized neighbor-joining phylogeny of Caprinae DRB exon 2 sequences from 10 species: domestic sheep (Ovar), bighorn sheep (Ovca), domestic and wild goat (Cahi, Caae, Capy), Ovibos moschatus (Ovmo), argali, mufflon, alpine chamois (Ruru, Schaschl et al., 2004) and Pyrenean chamois (Rupy, this work and Schaschl et al., 2005). For other references see Jugo and Vicario (2000). The rate used for dating divergence times is 0.97 × 10−9 substitutions per site per year (Satta et al., 1991).
Discussion
Our analysis of the second exon of the DRB1 gene of the MHC region has detected low levels of genetic diversity in the Iberian chamois, with a total of only 8 alleles in 130 animals, 7 alleles in R. p. pyrenaica and 3 alleles in R. p. parva. The lower number of alleles of R. p. parva remained after applying the rarefaction method, which standardizes the number of alleles to that of the smallest sample size. This number contrasts with other Caprinae such as R. rupicapra (19 alleles in 59 chamois) or bighorn sheep, where Gutierrez-Ezpeleta et al. (2001) identified a total of 21 alleles in 213 animals. On the other hand, a similar low value was found in the bottlenecked Spanish ibex, with only six different DRB1 alleles (Amills et al., 2004) and the endangered Arabian oryx (Hedrick et al., 2000) with three alleles.
Expected heterozygosity (He) was 0.609 in R. p. pyrenaica and slightly lower in R. p. parva, He=0.525. These values are quite similar to those observed in other Alpine species such as Capra pyrenaica victoriae and Capra pyrenaica hispanica, with values of 0.659 and 0.562, respectively (Amills et al., 2004). The total heterozygosity of R. rupicapra was higher (He=0.866) (Schaschl et al., 2004), but in this case simultaneous amplification of alleles from duplicated MHC loci could have inflated heterozygosity.
Two of the most common alleles in R. p. pyrenaica, Rupy-DRB1*02 and Rupy-DRB1*04, were also the most common alleles in R. p. parva (frequencies of 0.531 and 0.438) and, interestingly, these alleles were shared by R. pyrenaica and R. rupicapra (Schaschl et al., 2005). Such sharing of alleles is uncommon in ruminants, but might be explained by some form of conservative selection, as suggested by Doxiadis et al. (2006) to account for the pattern in rhesus and cynomolgus macaques, which share a large repertoire of identical Mhc class II exon 2 sequences.
It is estimated that Alpine and southern chamois split around 1.6+0.3 million years ago, based on divergence in the cytochrome b DNA sequence calibrated using the Myotragus subfossil (Lalueza-Fox et al., 2005). Among ruminants, this divergence time is similar to that between American bison and domestic cattle, which evolved from a common ancestor 1–1.5 million years ago (Loftus et al., 1994). Although the sequence motifs were virtually identical, none of the DRB alleles was shared between these species (Mikko et al., 1997; Traul et al., 2005), in contrast to our results.
The significant heterozygote deficiency (P<0.01) in the Aller population could be due to the presence of one or more nonamplifying null alleles, or because two different alleles have the same SSCP pattern. A second possibility is that the population is subdivided so that the heterozygosity is depressed by a Wahlund effect.
The extremely low MHC diversity of the parva subspecies could be the effect of one or more bottlenecks. In fact, this population has suffered several documented reductions, the most severe of them due to widespread hunting in the aftermath of the Spanish Civil War, when the population was reduced to fewer than 100 individuals (Nores and Vazquez, 1987), and more recently due to a sarcoptic mange outbreak that began in 1993 which reduced the population size by between 54 and 79% in some areas (Gonzalez-Quirós et al., 2002). Only two alleles were detected in the Cantabrian populations of Aller (n=20 animals) and Caso (n=6 animals). This number is even lower than the three alleles detected in the endangered oryx (Hedrick et al., 2000). Interestingly, these neighboring game reserves were affected by the sarcoptic mange epidemic (Fernández-Morán et al., 1997), but Somiedo was not. Recently bottlenecked populations are likely to have lost rare alleles, but may still contain substantial heterozygosity and quantitative genetic variation, which are lost more slowly than allelic variation (Luikart et al., 1998). Accordingly, no low-frequency alleles have been detected in the Cantabrian populations.
Although observed heterozygosity exceeded neutral expectations for most subpopulations, none showed a statistically significant excess of heterozygotes. On the other hand, amino acid diversity at functionally important sites indicates the strength of selection over evolutionary time scales. For R. pyrenaica, the dN/dS ratio for PBR across all DRB alleles is 7.23 (P<0.001). The number of replacement sites is roughly equally split between the α-helix and β-sheet, implying comparable selection in these two regions.
The phylogenetic trees revealed that the Pyrenean chamois alleles comprise at least three major allelic lineages, all of which were also present in the Alpine chamois. Their origin therefore appears to predate the divergence of the two species. In a comparison with DRB sequences of other Caprinae, chamois alleles form two monophyletic groups. Most of the alleles belong to one of the ancient lineages, exclusive to chamois, which may recently have split into two. The tree topology also suggests that most of the allelic diversity of the chamois lineages evolved after the split from other taxa. One possible explanation is a difference in the selective environment for these two sister species.
References
Amills M, Francino O, Sanchez A (1995). Nested PCR allows the characterization of Taq I and Pst I RFLPs in the second exon of the caprine MHC class II DRB gene. Vet Immunol Immunopathol 48: 313–321.
Amills M, Jimenez N, Jordana J, Riccardi A, Fernández-Arias A, Guiral J et al. (2004). Low diversity in the major histocompatibility complex class II DRB1 gene of the Spanish ibex, Capra pyrenaica. Heredity 93: 266–272.
Arnal MC, Fernández de Luco D (2004). Sanitary survey of Pyrenean chamois in the Aragonian Pyrenees (1997–2002). In: El sarrio pirenaico (ed). Rupicapra p. pyrenaica: Biología, patología y gestión. Consejo de protección de la Naturalez de Aragón y Fedenca: Zaragoza. pp 125–132.
Arrieta-Aguirre I, García-Etxebarria K, Jugo BM (2006). Optimization of MhcOvar-DRB1 typing. Tissue Antigens 67: 222–228.
Brown JH, Jardetzky TS, Gorga JC, Stern LJ, Urban RG, Strominger JL et al. (1993). Three-dimensional structure of human class II histocompatibility antigen HLA-DR. Nature 364: 33–39.
Charbonnel N, Pemberton J (2005). A long-term genetic survey of an ungulate population reveals balancing selection acting on MHC through spatial and temporal fluctuations in selection. Heredity 95: 1–12.
Doxiadis GG, Rouweler AJ, de Groot NG, Louwerse A, Otting N, Verschoor EJ et al. (2006). Extensive sharing of MHC class II alleles between rhesus and cynomolgus macaques. Immunogenetics 58: 259–268.
Ellegren H, Hartman G, Johansson M, Andersson L (1993). Major histocompatibility complex monomorphism and low levels of DNA fingerprinting variability in a reintroduced and rapidly expanding population of beavers. Proc Natl Acad Sci USA 90: 8150–8153.
Fernández-Morán J, Gómez S, Ballesteros F, Quirós P, Benito JL, Feliu C et al. (1997). Epizootiology of sarcoptic mange in a population of Cantabrian chamois (Rupicapra pyrenaica parva) in Northwestern Spain. Vet Parasitol 73: 163–171.
Gonzalez-Quirós P, Silva P, Solano S (2002). Population evolution of Cantabrian chamois Rupicapra pyrenaica parva with sarcoptic mange sarcoptes scabiei in Central-Eastern Asturias (Northwestern Spain). III World Congress on Mountain Ungulates. Saragosa, Spain, 35pp.
Guindon S, Gascuel O (2003). A simple, fast and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52: 696–704.
Gutierrez-Ezpeleta GA, Hedrick PW, Kalinowski ST, Garrigan D, Boyce WM (2001). Is the decline of desert bighorn sheep from infectious disease the result of low MHC variation? Heredity 86: 439–450.
Hall TA (1999). A Bioedit: a user friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucl Acids Symp Ser 41: 95–98.
Hedrick PW, Parker KM, Gutierrez-Espeleta GA, Rattink A, Lievers K (2000). Major histocompatibility complex variation in the Arabian oryx. Evolution Int J Org Evolution 54: 2145–2151.
Herrero J, Escudero E, García JM, García-Serrano A, Prada C, Couto S (2004). Management and survey on Pyrenean chamois in the Aragonian Pyrenees. In: El sarrio pirenaico (ed). Rupicapra p. pyrenaica: Biología, patología y gestión. Consejo de protección de la Naturalez de Aragón y Fedenca: Zaragoza. pp 69–82.
Hurlbert SH (1971). The nonconcept of species diversity: a critique and alternative parameters. Ecology 52: 577–586.
Jugo BM, Vicario A (2000). Single-strand conformational plymorphism and sequence polymorphism of Mhc-DRB in Latxa and Karrantzar sheep, implications for Caprinae phylogeny. Immunogenetics 51: 887–897.
Jukes TH, Cantor CR (1969). Evolution of protein molecules. In: Munroe HN (ed). Mammalian Protein Metabolism, Vol. 3. Academic Press: New York.
Klein J, Bontrop RE, Dawkins RL, Erlich HA, Gyllesten UB, Heise ER et al. (1990). Nomenclature for the major histocompatibility complexes of different species, a proposal. Immunogenetics 31: 217–219.
Konnai S, Nagaoka Y, Takeshima S, Onuma M, Aida Y (2003). Sequences and diversity of 17 new Ovar-DRB1 alleles from three breeds of sheep. Eur J Immunogenet 30: 275–282.
Kumar S, Tamura K, Nei M (2004). MEGA3: integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief Bioinform 5: 150–163.
Lalueza-Fox C, Castresana J, Sampietro L, Marques-Bonet T, Alcocer JA, Bertranpetit J (2005). Molecular dating of caprines using ancient DNA sequences of Myotragus balearicus, an extinct endemic Balearic mammal. BMC Evol Biol 6: 5–70.
Langella O (2002). Populations, version 1.2.28. CNRS, http://www.pge.cnrs-gif.fr/bioinfo/populations/index.php?lang=fr&doc=populations.
Loftus RT, McHugh DE, Bradley DG, Sharp PM, Cunningham P (1994). Evidence for two independent domestications of cattle. Proc Natl Acad Sci USA 91: 2757–2761.
Luikart G, Sherwin WB, Steele BM, Allendorf FW (1998). Usefulness of molecular markers for detecting population bottlenecks via monitoring genetic change. Mol Ecol 7: 963–974.
Masini F, Lovari S (1988). Systematics, phylogenetic relationship and dispersal of the chamois (Rupicapra spp.). Quaternary Res 30: 339–349.
Mikko S, Andersson L (1995). Low major histocompatibility complex class II diversity in European and North American moose. Proc Natl Acad Sci USA 92: 4259–4263.
Mikko S, Roed K, Schmutz S, Andersson L (1999). Monomorphism and polymorphism at Mhc DRB loci in domestic and wild ruminants. Immunol Rev Feb; 167: 169–178.
Mikko S, Spencer M, Morris B, Stabile S, Basu T, Stormont C et al. (1997). A comparative analysis of Mhc DRB3 polymorphism in the American Bison (Bison bison). J Hered 88: 499–503.
Nei M, Gojobori T (1986). Simple methods for estimating the numbers of synonymous and nonsynonimous nucleotide substitutions. Mol Biol Evol 3: 418–426.
Nei M, Tajima F, Tateno Y (1983). Accuracy of estimated phylogenetic trees from molecular data. II. Gene frequency data. J Mol Evol 91: 153–170.
Nores C, Vazquez VM (1987). La conservación de los vertebrados terrestres asturianos. Ministerio de Obras Públicas y Urbanismo, Centro de Publicaciones: Madrid, Spain (in Spanish).
O'Brien SJ, Evermann JF (1988). Interactive influence of infectious disease and genetic diversity in natural populations. Trends Ecol Evol 3: 254–259.
Orita M, Iwahana H, Kanazawa H, Hayashy K, Sekiya T (1989). Detection of polymorphism of human DNA by gel electrophoresis as single-strand conformation polymorphisms. Proc Natl Acad Sci USA 86: 2766–2770.
Pérez T, Albornoz J, Domínguez A (2002). Phylogeography of chamois (Rupicapra spp) inferred from microsatellites. Mol Phylogenet Evol 25: 524–534.
Petit RJ, El Mousadik A, Pons O (1998). Identifying populations for conservation on the basis of genetic markers. Conserv Biol 12: 844–855.
Potts WK, Wakeland EK (1990). Evolution of diversity of the major histocompatibility complex. Trends Ecol Evol 5: 181–187.
Raymond M, Rousset F (1995). GENEPOP (version 12), population genetics software for exact tests and ecumeniscism. J Hered 86: 248–249.
Sambrook J, Fritsch EF, Maniatis T (1989). Molecular Cloning. A Laboratory Manual, 2nd edn. Cold Spring Harbor Laboratory Press: New York.
Satta Y, Takahata N, Schönbach C, Gutknecht J, Klein J (1991). Calibrating evolutionary rates at major histocompatibility complex loci. In: Klein J, Klein D (eds). Molecular Evolution of the Major Histocompatibility Complex. Heidelberg: Springer. pp 51–62.
Schaschl H, Goodman SJ, Suchentrunk F (2004). Sequence analysis of the MHC class II DRB alleles in Alpine chamois (Rupicapra r. rupicapra). Dev Comp Immunol 91: 125–135.
Schaschl H, Suchentrunk F, Hammer S, Goodman SJ (2005). Recombination and the origin of sequence diversity in the DRB MHC Class II locus in chamois (Rupicapra sp.). Immunogenetics 57: 108–115.
Swofford DL (1998). Phylogenetic Analysis Using Parsimony (PAUP). University of Illinois: Champaign.
Takezaki N, Rzhetsky A, Nei M (1995). Phylogenetic test of the molecular clock and linearized trees. Mol Biol Evol 12: 823–833.
Traul DL, Bhushan B, Eldridge JA, Crawford TB, Li H, Davies CJ (2005). Characterization of Bison bison major histocompatibility complex class IIa haplotypes. Immunogenetics 57: 845–854.
Yuhki N, O'Brien SJ (1990). DNA variation at the mammalian major histocompatibility complex reflects genomic diversity and population history. Proc Natl Acad Sci USA 87: 836–840.
Acknowledgements
We thank Dr A Domínguez for providing us with Cantabrian chamois samples. We also thank Inés Arrieta-Agirre for her technical contribution in the SSCP analysis. Many thanks are due to hunters and gamekeepers that collected samples in the various sampling areas. This work was partially funded by Grant RTA02-003-C2-2 from MCyT to BMJ. Finally, we thank the anonymous reviewers for suggestions on the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Alvarez-Busto, J., García-Etxebarria, K., Herrero, J. et al. Diversity and evolution of the Mhc-DRB1 gene in the two endemic Iberian subspecies of Pyrenean chamois, Rupicapra pyrenaica. Heredity 99, 406–413 (2007). https://doi.org/10.1038/sj.hdy.6801016
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.hdy.6801016
Keywords
This article is cited by
-
Y-chromosome phylogeny in the evolutionary net of chamois (genus Rupicapra)
BMC Evolutionary Biology (2011)
-
Integrating phylogeographic patterns of microsatellite and mtDNA divergence to infer the evolutionary history of chamois (genus Rupicapra)
BMC Evolutionary Biology (2010)
