
Abstract
Strong evidence exists for global declines in pollinator populations. Data on the population genetics of solitary bees, especially diet specialists, are generally lacking. We studied the population genetics of the oligolectic bee Lasioglossum oenotherae, a specialist on the pollen of evening primrose (Onagraceae), by genotyping 455 females from 15 populations across the bee's North American range at six hyper-variable microsatellite loci. We found significant levels of genetic differentiation between populations, even at small geographic scales, as well as significant patterns of isolation by distance. However, using multilocus genotype assignment tests, we detected 11 first-generation migrants indicating that L. oenotherae's sub-populations are experiencing ongoing gene flow. Southern populations of L. oenotherae were significantly more likely to deviate from Hardy–Weinberg equilibrium and from genotypic equilibrium, suggesting regional differences in gene flow and/or drift and inbreeding. Short-term Ne estimated using temporal changes in allele frequencies in several populations ranged from ∼223 to 960. We discuss our findings in terms of the conservation genetics of specialist pollinators, a group of considerable ecological importance.
Similar content being viewed by others


Introduction
There is mounting evidence that pollinator assemblages, especially those of bees (Hymenoptera: Apoidea), are experiencing global declines, and this is expected to have severe consequences for both agricultural and natural systems (Biesmeijer et al., 2006). Furthermore, diet-specialist insects in general (Labandeira et al., 2002; Kotiaho et al., 2005), and bees in particular (Biesmeijer et al., 2006; Cane et al., 2006), seem to suffer higher extirpation rates when compared to generalists. For example, Cane et al. (2006) found that habitat fragmentation reduced the abundance and richness of oligolectic (diet specialist) but not polylectic (diet generalist) bees, suggesting higher extirpation rates in the former. This pattern was not found in one cavity-nesting specialist bee, but this is likely due to increased nesting opportunities in the urban matrix surrounding the fragments (Cane et al., 2006). The basis of higher extirpation rates for specialists is not well understood.
Since oligolectic bees make up a significant proportion of the world's bee fauna (Minckley and Roulston, 2006), a better knowledge of their conservation biology is therefore needed if we are to succeed in conserving the global bee fauna and other components of the biota that rely on their activities. However, population genetic studies of specialist pollinators have seldom been undertaken (Packer and Owen, 2001). Such studies are needed to examine how specialization affects population genetic parameters relevant to conservation (Packer et al., 2005; Zayed et al., 2005) and for testing hypotheses regarding the evolution of specialization and ecological speciation (Futuyma and Moreno, 1988; Funk et al., 2002), in addition to shedding light on the peculiar global patterns of bee biodiversity (Danforth et al., 2003).
In addition to extrinsic reasons, specialization can theoretically increase intrinsic extinction risk; as a consequence of specialization, the regional and local geographic distribution of oligolectic bees will be limited by that of their floral host(s), while polylectic bees should be able to maintain higher levels of gene flow through areas in which the host of a specialist species is absent. Thus, all other factors being equal, populations of specialists are expected to be more isolated than those of generalists (Futuyma and Moreno, 1988; Packer et al., 2005; Zayed et al., 2005). In addition, specialization can drive ecological divergence between specialist populations, through pre-mating isolation, which should also restrict gene flow. This can occur, for example, if specialists show strong site fidelity, or exhibit behaviors that enforce positive assortative mating (Funk et al., 2002). Further, if the development schedule of specialists is highly synchronized with their host-plant, as found in bees (Danforth, 1999; Minckley et al., 2000), then geographic variation in host-plant phenology can restrict gene flow between specialist populations (Linsley, 1958; Funk et al., 2002). These reductions in gene flow theoretically reduce effective population size (Ne) (Hedrick and Gilpin, 1997). Therefore, specialists are expected to suffer from higher extinction rates due to the intrinsic genetic problems of small populations (Frankham et al., 2002). Specialist bees are especially at risk given that, as is the case with other organisms with single locus complementary sex determination, they suffer from an extreme genetic load in small populations (Zayed and Packer, 2005). This occurs since loss of allelic richness at the sex locus leads to higher frequencies of homozygotes which mostly develop into inviable or effectively sterile diploid males instead of females (van Wilgenburg et al., 2006), leading to reductions in effective breeding population sizes (Zayed, 2004), and high rates of extinction that far surpass that caused by inbreeding depression in diploid animals (Zayed and Packer, 2005).
Only three studies on the population genetics of oligolectic bees have been undertaken. Using allozyme loci, Packer et al. (2005) found significantly reduced levels of genetic variation in the oligolectic member of five phylogenetically independent oligolectic – polylectic species pairs. Zayed et al. (2005), also using allozymes, found that populations of the specialist bee Leioproctus rufiventris were more genetically differentiated than those of the generalist Colletes seminitidus sampled from identical localities in the southern Atacama Desert. Finally, using microsatellite markers, Danforth et al. (2003) found high levels of genetic differentiation in the oligolectic bee Macrotera portalis in mixed Chihuahuan desert habitats in southeastern Arizona, USA. The above studies are consistent with the hypothesis that specialists persist in more isolated populations.
Here we report on the population genetics of an oligolectic solitary sweat bee, Lasioglossum (Sphecodogastra) oenotherae (Stevens), found in temperate eastern North America. The subgenus Sphecodogastra Ashmead (Halictidae: Lasioglossum) consists of eight species all of which specialize on pollen from evening primroses (Onagraceae) (McGinley, 2003). Females of Sphecodogastra have a unique scopa (that is, pollen collecting hairs) that is reduced to a single linear row of simple curved hairs on the hind femora, presumably an adaptation for collecting Onagraceae pollen that is held together by viscin threads (Knerer and MacKay, 1969; McGinley, 2003). Further, females forage for pollen either at dawn and/or dusk, or at night, which is rather unusual among bees (Knerer and MacKay, 1969; McGinley, 2003). L. oenotherae is found solely in eastern North America, from Georgia to the New England states in the USA, and from Ontario to New Brunswick in Canada, with four disjunct locality records in Kansas, Louisiana, Texas and North Dakota (McGinley, 2003).
The main purpose of our study is to examine the population genetics of the specialist L. oenotherae across its range in North America using recently isolated hyper-variable microsatellite markers (Zayed, 2006). Our study will also provide estimates for key parameters that are rarely known in solitary bee populations but are highly relevant to the conservation of these ecologically and economically important organisms.
Methods
Sampling
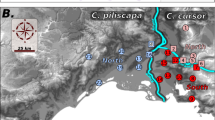
We obtained 15 L. oenotherae samples from eastern North America (Table 1; Figure 1), including a new record from the province of Quebec. One of us (AZ) spent extensive time (∼1 month) searching for L. oenotherae in Georgia and South Carolina in locations where it has been previously collected (McGinley, 2003). Although L. oenotherae's host plant was found blooming in abundance in the above-mentioned localities, we did not observe the bee foraging at dawn, dusk and during the day. Given that as a specialist bee, the emergence of L. oenotherae should coincide with that of its host's blooming period (Knerer and MacKay, 1969; McGinley, 2003), we take the absence of this bee in Georgia and South Carolina as anecdotal evidence of potential high extirpation rates at the southernmost parts of the bee's distribution.

Map of Lasioglossum oenotherae populations sampled for this study. Populations are indicated by black circles, and the numbers correspond to the last digit(s) of the population ID (see Table 1).
All of our collections were made on sundrops (mostly Oenothera fruticosa and O. pilosella) grown in gardens as we did not find this bee on ‘naturally’ occurring Oenothera. All populations were sampled during June and July 2005; however, we also sampled four populations during June and July 2001 (Table 1) to estimate short-term effective population sizes (Wang and Whitlock, 2003). We sampled females as they foraged on flowers during their peak flight activity (∼0700–0830 hours). Females were killed in the field by immersion in liquid nitrogen (or in a few cases, using 95% ethanol) until transport to York University where samples were stored at −80°C until further processing.
DNA extraction and polymerase chain reaction
We extracted genomic DNA from L. oenotherae thoraces using a GenElute Mammalian Genomic DNA Miniprep Kit (Sigma, St Louis, MA, USA) following the manufacture's protocols (including the RNAse treatment). The extracted DNA was eluted in 200 μl of the supplied elution buffer. The remainder of the bee was stored as a voucher at −80°C. After extractions, we genotyped the bees at the following six variable microsatellite loci (Zayed, 2006): A31, A33, C8, D5, D53 and D71. Loci D53, D71 and A33 were amplified using fluorescently labeled forward primers with D2, D2 and D3 WellRed dyes (IDT), respectively, in a 10 μl polymerase chain reaction (PCR) containing 20–40 ng DNA, 0.3 μ M labeled-forward and -reverse primers each, 0.2 mM of each dNTP (Fermentas, Hanover, MD, USA), 1 × Tsg buffer, 2.2 mM MgSO4 and 0.2 U Tsg DNA polymerase (Biobasic, Markham, ON, Canada). The reactions were performed in a Mastercycler gradient thermocycler (Eppendorf, Hamburg, Germany) with the following conditions: 94°C for 5 min; 30 cycles at 94°C for 30 s, 60°C for 45 s, 72°C for 45 s and a final extension at 72°C for 4 min holding at 4°C. The remaining loci were amplified in a nested PCR with a fluorescently labeled universal primer (Zayed, 2006) in a 10 μl reaction containing 20–40 ng DNA, 0.3 μ M reverse primer, 0.075 μ M forward primer with a 5′ M13 tail, 0.3 μ M M13(−21) primer labeled with either D3 (for A31) or D4 (for C8 and D5) WellRed dyes (IDT), 0.2 mM of each dNTP (Fermentas), 1 μl bovine serum albumin (250 μg/ml, Sigma), 1 × Tsg buffer, 2.2 mM MgSO4 and 0.2 U Tsg DNA polymerase (Biobasic). Cycling conditions are: 94°C for 5 min, 30 cycles at 94°C for 30 s, 60°C for 45 s, 72°C for 45 s; 8 cycles at 94°C for 30 s, 53°C for 45 s, 72°C for 45 s and a final extension at 72°C for 10 min holding at 4°C.
Allele sizes for loci labeled with the same dye did not overlap, and thus it was possible to poolplex products from all six loci in the following ratio: 3.8 μl D53 (D2):2 μl D71 (D2):0.92 μl A31 (D3):0.6 μl A33 (D3):0.4 μl C8 (D4):0.75 μl D5 (D4). Fragment analysis was subsequently performed on a CEQ 8000 (Beckman Coulter, Fullerton, CA, USA) in a 40 μl reaction containing 1 μl of the poolplex solution, 38.5 μl sampling loading solution and 0.5 μl 400 bp size standard (Beckman Coulter).
Data analysis
We used exact Hardy–Weinberg equilibrium (HWE) tests, as implemented in GENEPOP version 3.3 (Raymond and Rousset, 1995), to examine if loci (over all populations) or populations (over all loci) significantly deviated from HWE. We examined genotypic disequilibrium between loci (GD) (that is nonrandom association of genotypes between loci) over all populations using randomization tests as implemented in FSTAT version 2.9.3 (Goudet, 1995). Significance of the HWE and GD tests was assessed following standard Bonferroni corrections (Rice, 1989). Based on geographic proximately, and a genetic distance phenogram (see Methods and Results), we conducted the above tests for the entire data set, and for subsets containing populations north of Long Island (5SO9–5SO15, hereafter called northern populations), and south of and including Long Island (5SO1–5SO8, hereafter called southern populations). We used χ2-tests (Zar, 1999) to examine differences in the occurrence of significant deviations from HWE and GD between northern and southern populations – for this analysis, we counted tests as significant before Bonferroni corrections (P<0.05).
We quantified genetic diversity using Nei's unbiased expected heterozygosity Hexp (Nei, 1978) and allelic richness NA corrected for sample size (El Mousadik and Petit, 1996), as estimated in FSTAT. We used a Kruskal–Wallis test (Zar, 1999) to examine if genetic diversity was equal among our populations. We also used the same test to examine if mean Hexp and NA significantly differed between northern and southern populations of L. oenotherae.
We used hierarchical F-statistics to examine the extent of population structure in L. oenotherae. We estimated global F-statistics (FIT, FST and FIS) and pairwise FST following Cockerham and Weir (1993), as implemented in FSTAT (Goudet, 1995), and the significance of these estimates was determined using 1000 randomizations. Ninety-five percent confidence intervals for the F-statistics were estimated by bootstrapping over loci (Goudet, 1995). We estimated GST′, a standardized measure of global genetic differentiation which is independent of the amount of genetic variation observed at the examined loci, to facilitate comparisons with future studies (Hedrick, 2005). GST′ was estimated using MSA 4.05 (Dieringer and Schlötterer, 2003), and its significance was estimated using 1000 permutations. We also examined the population genetic structure using a hierarchical analysis of molecular variance (AMOVA) framework (Excoffier et al., 1992), as implemented in ARLEQUIN 3.01 (Excoffier et al., 2005), which attempts to partition the total variance in gene frequencies into components due to the following sources of structure: among geographic regions (north versus south), among populations within the geographic regions and within populations. We also estimated Nei's genetic distance between all population pairs (Nei, 1978) using GENALEX (Peakall and Smouse, 2006), and used the estimates to construct an UPGMA phenogram using the subroutine NEIGHBOR, and DRAWGRAM in PHYLIP v3.6 (Felsenstein, 1989). We conducted isolation by distance analyses (IBD) by regressing pairwise FST, as well as Nei's genetic distance, against geographic distance using linear regression as implemented in GENALEX. The significance of IBD relationships was examined using a Mantel test with 999 permutation using GENALEX.
Additionally, we carried out Bayesian population structure analysis using STRUCTURE v2.1 (Pritchard et al., 2000; Falush et al., 2003) and GENELAND v1.0.8 (Guillot et al., 2005). Loosely described, both packages attempt to infer the number of populations present in the data set, where different populations have distinct allele frequencies and are assumed to be at HWE. GENELAND explicitly uses the spatial coordinates (that is, latitude and longitude) of the sampled individuals to assist in clustering the samples, while STRUCTURE can implicitly include spatial information (that is, the site where individuals were collected) to assist clustering. For both programs, we used a subset of bees (N=306) which had no missing genotypes sampled during 2005. For STRUCTURE, we conducted our analysis using the prior population information model to assist clustering, using the default parameters, running the simulations for values of K from 2 to 15 populations, and for each value of K, we set the burnin period to 50 000 repetitions followed by 500 000 MCMC repetitions. GENELAND analyses were also conducted for values of K ranging from 2 to 15 populations using the spatial Dirichlet model with 500 000 MCMC iterations (other parameters, such as ‘rate max’ and ‘nb.Nuclei max’, were estimated based on the number of individuals in our sample as per the authors' instructions). We would like to note that the number of loci used in this study is below the number recommended (<10) by both STRUCTURE and GENELAND, and lack of HWE and linkage equilibrium (see Results) in many of the sampled populations are in violation of both programs' assumptions (Pritchard et al., 2000; Falush et al., 2003; Guillot et al., 2005). Further, GENELAND assumes that individuals used for clustering were randomly sampled across the study area – an assumption which is also violated in our study (Guillot et al., 2005). The consequences of violating these assumptions are unknown, and thus we urge the readers to take the results of the Bayesian population structure analysis with a grain of salt.
To examine the extent of current gene flow between L. oenotherae populations, we used multilocus genotype assignment tests to detect first-generation migrants as implemented in GENECLASS2 (Piry et al., 2004). A first-generation migrant is defined as a bee that was not born into the population from which it was sampled. We used Rannala and Mountain's (1997) method for estimating the likelihood of multilocus genotypes of all individuals in all populations. To generate critical values to test the null hypothesis of an individual being a resident in its sampled populations, we employed Monte-Carlo resampling simulations, where 1000 simulated individuals were created by drawing multilocus gametes with replacement from each population (Paetkau et al., 2004). To reduce type I errors, we evaluated the significance of our tests given α=0.01 as recommended by Paetkau et al. (2004).
In populations with samples from multiple generations, we attempted to estimate short-term effective population size, Ne, using a maximum likelihood temporal method implemented in MNE2 (Wang and Whitlock, 2003).
Results
Hardy–Weinberg and genotypic disequilibrium
Significant deviations from Hardy–Weinberg and genotypic equilibrium were detected in our data set. Over all populations, three loci significantly deviated from HWE: A33, D53 and D71, all exhibiting an excess of homozygotes. Five populations also significantly deviated from HWE over all loci, also showing an excess of homozygotes (Table 2). Out of 15 possible locus pairs, 4 pairs showed significant GD (C8 and D71, D53 and A33, D53 and C8, and D53 and D71), all involving either D53 or D71. However, the above patterns were driven by southern L. oenotherae populations. A χ2-test revealed that a larger number of southern populations significantly deviated from HWE when compared to northern populations (d.f.=1, χ2=8.04, P<0.01). Similarly, more locus pairs were significantly linked in southern versus northern populations (d.f.=1, χ2=9.6, P<0.01). Over all southern populations, A33, D53 and D71 continued to significantly deviate from HWE. Additionally, eight locus pairs showed significant GD (A31 and C8, A31 and D53, A31 and D71, A33 and D71, C8 and D53, D5 and D53, D5 and D71, and D53 and D71) in southern populations. However, in the northern populations, none of the loci were found to significantly deviate from HWE, and no GD was detected between any of the possible locus pairs. This evidence suggests that departures from HWE and genotypic equilibrium are not generated by null alleles or physical linkage of the studied loci, respectively, but are rather due to population genetic processes acting preferentially on the southern L. oenotherae populations. However, we still investigated the consequences of GD in our data set by conducting our population structure analyses with all loci and with D53 and D71 removed. The latter only caused slight changes in parameter estimates, but did not change the significance of our statistical tests, and thus we report results based on the analysis of all loci.
Genetic diversity and population structure
Genetic diversity significantly differed among L. oenotherae populations (Kruskal–Wallis test, P=0.026 for Hexp, P=0.026 for NA). Further, mean Hexp and NA were significantly lower in northern populations versus southern populations (Kruskal–Wallis test, P=0.0003 for Hexp, P=0.0007 for NA). The significance of our tests did not change when we excluded the population from Waterloo (5SO11) that had particularly low levels of genetic diversity (Table 2).
L. oenotherae populations showed significant levels of genetic differentiation. A significant excess of homozygosity over HWE was found over all populations (FIT=0.15, 95% confidence interval (CI): 0.10–0.19, P<0.001), mostly due to population subdivision (FST=0.10, 95% CI: 0.07–0.14, P<0.001) and, to a lesser extent, to nonrandom mating within local populations (FIS=0.05, 95% CI: 0.02–0.08, P<0.001). The standardized global genetic differentiation measure GST′=0.52 (P<0.001) also indicated significant levels of genetic differentiation. Out of 105 estimates of pairwise FST, 102 estimates were significant after Bonferroni corrections (all tests were significant before corrections) indicating that nearly all populations were genetically differentiated (Table 3). We also detected regional differences in levels of population structure, with an average pairwise FST=0.094 between northern populations, and an average pairwise FST=0.054 between southern populations, indicating that northern populations are more genetically differentiated. Hierarchical AMOVA analysis also detected significant levels of structure among regions (North versus South), among populations within regions and among all populations (Table 4).
The results of the Bayesian population structure analyses also indicate that L. oenotherae exhibits significant genetic differentiation across its range in North America. Using STRUCTURE, a model with K=15 populations received the highest likelihood, and each one of our samples was assigned as a unique population. Using GENELAND, we found that a model with K=7 populations best fit our data: cluster 1 included 5SO9, 12 and 13 (average pairwise distance=193 km, FST=0.07); cluster 2 included 5S010, 11, 14 and 15 (average pairwise distance=221 km, FST=0.083); cluster 3 included the four samples collected on long island (5SO5–8, average pairwise distance=60 km, FST=0.03); cluster 4 included 5SO1 and 2 (average pairwise distance=236 km, FST=0.04), while 5SO3 and 5SO4 clustered as separate populations (refer to map in Figure 1). The usually large geographic distances and significant FST estimates between samples contained in multi-sample clusters were surprising. Nevertheless, sequential GENELAND analysis on the multi-sample clusters determined from the first run managed to uncover differences between almost all of our samples. For example, subsequent analysis on a subset of the data containing only northern samples eventually recovered all populations as being distinct, with the exception of 5SO12 and 5SO13, which do have a very small and insignificant pariwise FST value (Table 3).
We detected a significant pattern of isolation by distance between L. oenotherae populations evident from both the significant correlation between pairwise FST and geographic distance as well as between Nei's genetic distance and geographic distance (Figure 2; Mantel: P<0.01 for both tests). A phenogram based on Nei's pairwise genetic distance (Figure 3) confirms both the extent of population structure and IBD in L. oenotherae populations. Using multilocus assignment analyses, we found that most bees were assigned high probabilities of being residents of (that is born into) the population from which they were sampled. Out of 357 bees sampled in 2005, 11 were identified as first-generation immigrants in the following populations: 5SO1, 5SO3, 5SO4, 5SO6, 5SO7 (two immigrants), 5SO8, 5SO9, 5SO10, 5SO13 and 5SO15.

Isolation by distance between L. oenotherae populations. (a) Pairwise estimates of genetic differentiation, as well as (b) estimates of genetic distance were both significantly positively correlated with geographic distance.

A phenogram depicting genetic distances between Lasioglossum oenotherae populations.
Maximum likelihood estimates of short-term effective population size were obtained for three L. oenotherae populations sampled over multiple generations: Watkins Glen (NY, SO9) – Ne=335.39 (95% CI: 83.16–NA), Ithaca (NY, SO10) – Ne=223.27 (95% CI: 85.93–NA) and Toronto (ON, SO12) – Ne=960.99 (95% CI: 124.43–NA). The upper 95% interval for these values could not be estimated with confidence, likely due to a combination of too little genetic differentiation between generations and the number of loci examined (Wang and Whitlock, 2003). Similarly, the Ne of the Waterloo population (SO11) could not be estimated with any confidence.
Discussion
Population structure of specialists
We found significant levels of population structure in L. oenotherae sampled from across its range in North America (Global FST=0.10, GST′=0.52). Significant levels of genetic differentiation were detected between almost all sampled populations even at distances of <10 km (Table 3). Bayesian analyses also supported the extent of structure in L. oenotherae populations, although one model (GENELAND) experienced some difficulties with initially detecting structure between some of the sampled populations. This may be due to violations of GENELAND's assumptions (see Methods) or the low number of loci used in our data set. Nevertheless, GENELAND was able to recover almost all of our samples as distinct after sequential analysis. We also detected a significant relationship between genetic differentiation/genetic distance, and geographic distance in L. oenotherae populations (Figure 2). A phenogram based on Nei's genetic distance also confirms a general relationship between geographic and genetic distances (Figure 3). The two major groups on the phenogram correspond to our defined groups of northern and southern populations. Using multilocus genotype assignment analyses, we detected several first-generation migrants, indicating the L. oenotherae populations experience ongoing gene flow, thereby reducing the rate at which they diverge due to drift and/or local adaptation.
Significant population structure is expected in specialists (reviewed by Futuyma and Moreno, 1988; Funk et al., 2002; Packer et al., 2005; Zayed et al., 2005), as found for L. oenotherae. Until now, only two studies of the population structure of specialist bees have been undertaken, with both studies documenting significant structure in specialist bees in xeric environments (Danforth et al., 2003; Zayed et al., 2005). Our study on L. oenotherae in temperate eastern North America thus suggests that significant population structure may be common to all specialist bees. A number of comparative studies have recently shown that specialist populations tend to be more genetically differentiated when compared to closely related generalists (bark beetles – Kelley et al., 2000; dove lice – Johnson et al., 2002; aphids – Gaete-Eastman et al., 2004), supporting the hypothesis that specialization promotes isolation (Futuyma and Moreno, 1988; Funk et al., 2002). Although the population genetics of L. oenotherae are consistent with the aforementioned hypothesis, it is difficult to conclude if specialization is responsible for the significant population structure without comparative data to eliminate other possibly confounding effects. The global FST estimate in L. oenotherae populations ranked in the highest 25% (20/95) of insects surveyed for that parameter (data from Peterson and Denno, 1998); however, the meta-analysis included only seven hymenopteran species. Given that FST estimates are dependent on the underlying levels of genetic variation at the studied loci (Hedrick, 2005), it is also difficult to compare our results to other studies using FST. Comparing our results for L. oenotherae to other population genetic studies of bees utilizing microsatellite markers will likely provide a less biased perspective. Unfortunately, most bee population genetic studies involve social species, confounding sociality with host plant breadth in any comparison. Further, studies on social species often involve a single population (Paxton et al., 2000), preventing analysis of population structure. More population genetic data on solitary generalist Lasioglossum in particular, and solitary bees in general, are therefore needed to examine the consequences of diet breadth on the population genetics of specialist pollinators.
Effective population size
Short-term Ne was large for three L. oenotherae populations sampled from multiple generations (∼220–960). This is somewhat surprising given that specialists are expected to persist in populations with smaller effective sizes (Packer et al., 2005), and also given other previous estimates of Ne in generalist bees (Zayed and Packer, 2001; Zayed, 2004). However, previous estimates were based on equilibrium models using allozyme data and reflect historical Ne, rather than short-term Ne. Further, all our samples were from gardens where Oenothera densities may be kept artificially high, allowing for large bee populations. Such garden populations may also be artificially sheltered from yearly fluctuations in size, which would otherwise decrease Ne (Frankham et al., 2002). It is interesting to note that the highest Ne estimate was obtained from the largest city, which is presumably reflective of larger Oenothera populations in Toronto versus Watkins Glen and Ithaca.
HWE and GD in southern populations
Southern populations significantly deviated from HWE and genotypic equilibrium when compared to northern populations. The geographic pattern of these deviations and the number of loci involved strongly suggest that this pattern is not generated by null alleles or actual linkage between loci. Ignoring selection and mutation, GD can be generated by either inbreeding, drift in small populations or by migration between structured populations (Nei and Li, 1973; Hedrick, 2000). Inbreeding increases homozygosity which reduces the effective recombination rate and increases genotypic disequilibrium, while drift can create nonrandom associations between alleles at different loci by chance events in small populations, which may persist for long periods when combined with inbreeding. Finally, migration can increase genotypic disequilibrium if two structured populations exchange migrants since the multilocus genotypes of immigrants and their descendents are likely to be sufficiently different from those of the original inhabitants (Nei and Li, 1973; Hedrick, 2000). Genetic evidence exists for the latter hypothesis, since the average pairwise FST of southern populations is nearly half of that found in northern populations, implying that southern populations exchange more migrants, and this can account for the differences in genotypic disequilibrium between the two regions.
Inbreeding and drift can also be responsible for the observed patterns of GD, which were geographically correlated with deviations from HWE in southern populations. As we noted in the methods, we could not find L. oenotherae populations previously recorded from the southernmost portions of its range, suggesting preferential extirpation of southern populations. If southern L. oenotherae populations are experiencing higher extirpation rates, then we would expect them to show some signatures of small population size, such as the observed homozygosity excess and GD. Strong bottlenecks are especially known to create high levels of GD between unlinked markers (Nordborg et al., 2002). However, higher levels of genetic diversity are found in southern populations when compared to northern populations, which is inconsistent with inbreeding and drift as explanations for GD and deviations from HWE. Finally, pooling data from multiple generations (that is, if we sampled southern populations at a time where both over-wintered mothers and their daughters were flying) can theoretically cause deviations in HWE (Tonsor et al., 1993) and genotypic disequilibrium if allele frequencies vary substantially from year to year. However, this is unlikely since we did not observe any males (that is sons) flying indicating that we only sampled over-wintered mothers (AZ personal observation). Further work is needed to untangle the possible effects of the above-mentioned factors on the observed deviations from HWE and GD.
Conclusions
Our study documents significant levels of genetic structure in a solitary specialist bee which is experiencing ongoing gene flow. Regional differences in deviations from HWE and GD in L. oenotherae populations were detected which warrant additional attention. Further studies on the population genetics of specialist and generalist bees are needed to examine the consequences of specialization on the population and conservation biology of specialist, a major component of the global bee fauna.
References
Biesmeijer JC, Roberts SPM, Reemer M, Ohlemuller R, Edwards M, Peeters T et al. (2006). Parallel declines in pollinators and insect-pollinated plants in Britain and the Netherlands. Science 313: 351–354.
Cane JH, Minckley RL, Kervin LJ, Roulston TH, Williams NM (2006). Complex responses within a desert bee guild (Hymenoptera: Apiformes) to urban habitat fragmentation. Ecol Appl 16: 632–644.
Cockerham CC, Weir BS (1993). Estimation of gene flow from F-statistics. Evolution 47: 855–863.
Danforth BN (1999). Emergence dynamics and bet hedging in a desert bee, Perdita portalis. Proc Biol Sci 266: 1985–1994.
Danforth BN, Ji S, Ballard LJ (2003). Gene flow and population structure in an oligolectic desert bee, Macrotera (Macroteropsis) portalis (Hymenoptera: Andrenidae). J Kans Entomol Soc 76: 221–235.
Dieringer D, Schlötterer C (2003). Microsatellite analyser (MSA): a platform independent analysis tool for large microsatellite data sets. Mol Ecol Notes 3: 167–169.
El Mousadik A, Petit RJ (1996). High level of genetic differentiation for allelic richness among populations of the argan tree [Argania spinosa (L.) Skeels] endemic to Morocco. Theor Appl Genet 92: 832–839.
Excoffier L, Laval G, Schneider S (2005). Arlequin ver. 3.0: an integrated software package for population genetics data analysis. Evol Bioinform Online 1: 47–50.
Excoffier L, Smouse PE, Quattro JM (1992). Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131: 479–491.
Falush D, Stephens M, Pritchard JK (2003). Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164: 1567–1587.
Felsenstein J (1989). PHYLIP – Phylogeny Inference Package (Version 3.2). Cladistics 5: 164–166.
Frankham R, Ballou JD, Briscoe DA (2002). Introduction to Conservation Genetics. Cambridge University Press: Cambridge.
Funk JD, Filchak KE, Feder JL (2002). Herbivorous insects: model systems for the comparative study of speciation ecology. Genetica 116: 251–267.
Futuyma DJ, Moreno G (1988). The evolution of ecological specialization. Annu Rev Ecol Syst 19: 207–233.
Gaete-Eastman C, Figueroa CC, Olivares-Donoso R, Niemeyer HM, Ramirez CC (2004). Diet breadth and its relationship with genetic diversity and differentiation: the case of southern beech aphids (Hemiptera: Aphididae). Bull Entomol Res 94: 219–227.
Goudet J (1995). FSTAT, version 1.2: a computer program to calculate F statistics. J Hered 86: 485–486.
Guillot G, Mortier F, Estoup A (2005). GENELAND: a computer package for landscape genetics. Mol Ecol Notes 5: 712–715.
Hedrick PW (2000). Genetics of Populations, 2nd edn. Jones and Bartlett Publishers: Sudbury, MA.
Hedrick PW (2005). A standardized genetic differentiation measure. Evolution 59: 1633–1638.
Hedrick PW, Gilpin ME (1997). Genetic effective size of a metapopulation. In: Hanski IA, Gilpin ME (eds). Metapopulation Biology: Ecology, Genetics, and Evolution. Academic Press Inc.: San Diego, USA. pp 165–181.
Johnson KP, Williams BL, Drown DM, Adams RJ, Clayton DH (2002). The population genetics of host specificity: genetic differentiation in dove lice (Insecta: Phthiraptera). Mol Ecol 11: 25–38.
Kelley ST, Farrell BD, Mitton JB (2000). Effect of specialization on genetic differentiation in sister species of bark beetles. Heredity 84: 218–227.
Knerer G, MacKay P (1969). Bionomic notes on the solitary Evylaeus oenotherae (Stevens) (Hymenoptera: Halictinae), a matinal summer bee visiting cultivated Onagraceae. Can J Zool 47: 289–294.
Kotiaho JS, Kaitala V, Komonen A, Päivinen J (2005). Predicting the risk of extinction from shared ecological characteristics. Proc Natl Acad Sci USA 102: 1963–1967.
Labandeira CC, Johnson KR, Wilf P (2002). Impact of the terminal Cretaceous event on plant-insect associations. Proc Natl Acad Sci USA 99: 2061–2066.
Linsley EG (1958). The ecology of solitary bees. Hilgardia 27: 543–599.
McGinley RJ (2003). Studies of Halictinae (Apoidea: Halictidae), II: revision of Sphecodogastra Ashmead, floral specialists of Onagraceae. Smithson Contrib Zool 610: 1–55.
Minckley RL, Cane JH, Kervin L (2000). Origins and ecological consequences of pollen specialization among desert bees. Proc Biol Sci 267: 265–271.
Minckley RL, Roulston TH (2006). Incidental mutualisms and pollen specialization among bees. In: Waser NM, Ollerton J (eds). Plant-Pollinator Interactions: From Specialization to Generalization. University of Chicago Press: Chicago, USA. pp 69–98.
Nei M (1978). Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89: 583–590.
Nei M, Li WH (1973). Linkage disequilibrium in subdivided populations. Genetics 75: 213–219.
Nordborg M, Borevitz JO, Bergelson J, Berry CC, Chory J, Hagenblad J et al. (2002). The extent of linkage disequilibrium in Arabidopsis thaliana. Nat Genet 30: 190–193.
Packer L, Owen R (2001). Population genetic aspects of pollinator decline. Conserv Ecol 5: 4 (online) URL: http://www.consecol.org/vol5/iss1/art.
Packer L, Zayed A, Grixti JC, Ruz L, Owen RE, Vivallo F et al. (2005). Conservation genetics of potentially endangered mutualisms: reduced levels of genetic variation in specialist versus generalist bees. Conserv Biol 19: 195–202.
Paetkau D, Slade R, Burden M, Estoup A (2004). Genetic assignment methods for the direct, real-time estimation of migration rate: a simulation-based exploration of accuracy and power. Mol Ecol 13: 55–65.
Paxton RJ, Thorén P, Gyllenstrand N (2000). Microsatellite DNA analysis reveals low diploid male production in a communal bee with inbreeding. Biol J Linn Soc Lond 69: 483–502.
Peakall R, Smouse PE (2006). GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6: 288–295.
Peterson MA, Denno RF (1998). Life history strategies and the genetic structure of phytophagous insect populations. In: Mopper S, Strauss SY (eds). Genetic Structure and Local Adaptation in Natural Insect Populations: Effects of Ecology, Life History and Behavior. Chapman & Hall: New York, USA. pp 263–322.
Piry S, Alapetite A, Cornuet JM, Paetkau D, Baudouin L, Estoup A (2004). GeneClass2: a software for genetic assignment and first-generation migrant detection. J Hered 95: 536–539.
Pritchard JK, Stephens M, Donnelly P (2000). Inference of population structure using multilocus genotype data. Genetics 155: 945–959.
Rannala B, Mountain JL (1997). Detecting immigration by using multilocus genotypes. Proc Natl Acad Sci USA 94: 9197–9221.
Raymond M, Rousset F (1995). GENEPOP (version 1.2): population genetics software for exact tests and ecumenicism. J Hered 86: 248–249.
Rice WR (1989). Analysing tables of statistical tests. Evolution 43: 223–225.
Tonsor SJ, KIalisz S, Fisher J, Holtsford TP (1993). A life-history based study of population genetic structure: see bank to adults in Plantago lanceolata. Evolution 47: 833–843.
van Wilgenburg E, Driessen G, Beukeboom L (2006). Single locus complementary sex determination in Hymenoptera: an ‘unintelligent’ design? Front Zool 3: 1–15.
Wang J, Whitlock MC (2003). Estimating effective population size and migration rates from genetic samples over space and time. Genetics 163: 429–446.
Zar JH (1999). Biostatistical Analysis. Prentice-Hall Inc.: Upper Saddle River, NJ.
Zayed A (2004). Effective population size in Hymenoptera with complementary sex determination. Heredity 93: 627–630.
Zayed A (2006). Characterization of microsatellite loci from the solitary sweat bees Lasioglossum leucozonium and Lasioglossum oenotherae (Hymenoptera, Halictidae). Mol Ecol Notes 6: 1154–1156.
Zayed A, Packer L (2001). High levels of diploid male production in a primitively eusocial bee (Hymenoptera: Halictidae). Heredity 87: 631–636.
Zayed A, Packer L (2005). Complementary sex determination substantially increases extinction proneness of haplodiploid populations. Proc Natl Acad Sci USA 102: 10742–10746.
Zayed A, Packer L, Grixti JC, Ruz L, Toro H, Owen RE (2005). Increased genetic differentiation in a specialist versus a generalist bee: implications for conservation. Conserv Genet 6: 1017–1026.
Acknowledgements
This work was funded by a Canadian Graduate Scholarship to AZ and Natural Science and Engineering Research Council of Canada grants to LP. We thank JC Grixti for assistance in sampling L. oenotherae. We also thank B Danforth, J Ascher and R Winfree for their help in locating bee populations, and AS Constantin and L Gibbens for laboratory assistance. JC Grixti, SI Wright and anonymous reviewers provided helpful comments on the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zayed, A., Packer, L. The population genetics of a solitary oligolectic sweat bee, Lasioglossum (Sphecodogastra) oenotherae (Hymenoptera: Halictidae). Heredity 99, 397–405 (2007). https://doi.org/10.1038/sj.hdy.6801013
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.hdy.6801013
Keywords
This article is cited by
-
Population assessment and foraging ecology of the rare solitary bee Anthophora retusa at Seaford Head Nature reserve
Journal of Insect Conservation (2021)
-
Lack of genetic structuring, low effective population sizes and major bottlenecks characterise common and German wasps in New Zealand
Biological Invasions (2019)
-
Uneven distribution of complementary sex determiner (csd) alleles in Apis mellifera population
Scientific Reports (2017)
-
Accessing the genetic content of Xylocopa frontalis bees (Apidae, Xylocopini) for sustainable management in pollination services of passion fruit
Apidologie (2017)
-
Mediterranean lineage endemism, cold-adapted palaeodemographic dynamics and recent changes in population size in two solitary bees of the genus Anthophora
Conservation Genetics (2017)
