
Abstract
The gametophytic self-incompatibility locus has been thought to be a nonrecombining genomic region. Inferences have been made, however, about the functional importance of different parts of the S-locus, based on differences in the levels of variability along the gene, and this is valid only if recombination occurs. It is thus important to test whether recombination occurs within and near the S-locus. Several recent attempts to test this have reached conflicting conclusions. In this study, we examine a large data set on sequence variation at the S-locus in several species with gametophytic self-incompatibility systems, in the Solanaceae, Rosaceae and Scrophulariaceae. We use the longest sequences available to test for recombination based on linkage disequilibrium between polymorphic sites in the S-locus. The relationship between linkage disequilibrium and physical distance between the sites suggests rare intragenic exchange in the evolutionary history of four species of Solanaceae and two species of Rosaceae.
Similar content being viewed by others


Introduction
The self-incompatibility system of many flowering plants ensures that pollen cannot fertilize a plant's own ovules. In most self-incompatible species, this is controlled by alleles at a single S-locus (de Nettancourt, 1977). S-alleles determining rare specificities have a reproductive advantage over alleles for common incompatibility types, and many different alleles are expected to be maintained at approximately equal frequencies for long periods of time, even in finite populations (Vekemans and Slatkin, 1994; Clark, 1996). Very high levels of amino-acid and silent-site polymorphism are thus expected, and observed, at the S-locus (Clark, 1993).
As alleles may persist for long periods of time, large sequence differences can develop if recombination between the S-alleles does not occur, or is very rare, consistent with the extreme differences found between allele sequences at incompatibility loci (eg Richman et al, 1996; Awadalla and Charlesworth, 1999; Richman and Kohn 2000; Vieira and Charlesworth, 2002). Hypervariability in certain regions of the S-locus has been taken as indicating parts of the gene that encode the regions of the stigmatic S protein involved in specificity differences (reviewed in Awadalla and Charlesworth, 1999). In the absence of recombination, sequence variants within a functional allelic class (ie sequences with the same specificity) will indeed be associated with that specificity until separated by recombination (Strobeck, 1972), unless recurrent mutation occurs at the same site. If only a few sites determine specificity differences, peaks of variability are expected in regions close to these sites (Nordborg et al, 1996), as in the MHC loci (Takahata and Satta, 1998a, 1998b).
However, identifying local peaks of variability in sequences as regions under balancing selection (ie recognition sequences) is valid only if recombination or gene conversion occur, separating sites under selection from associations that arise by mutation. Without such exchange, higher and lower variabilities can arise, due to differences in selective constraints, but the balanced polymorphism at the S-locus will increase variability throughout the gene, as is indeed observed for silent and intron sites as well as for nonsynonymous sites (Awadalla and Charlesworth, 1999; Schierup et al, 2001; Vieira and Charlesworth, 2002). It is therefore important to determine whether S-loci show recombination or not. If recombination occurs, the number of peaks in variability could also help distinguish whether balancing selection acts at many sites in the sequence, or at only a few sites.
Until recently, the gametophytic self-incompatibility locus was thought not to recombine (Clark, 1993). In two species of Solanaceae, Lycopersicon peruvianum (Bernatzky, 1993) and Petunia hybrida (Entani et al, 1999), the S-locus maps to the centromeric region, and the organization of this region is thought to be conserved in other species of Solanaceae (ten Hoopen et al, 1998). Centromeric regions have suppressed crossing over in a wide range of species, including plants (reviewed by Charlesworth and Charlesworth, 1998). The S-loci of these species may therefore be in a low-recombination region of the genome. In Rosaceae, however, the data suggest a noncentromeric localization of the S-locus (Ushijima et al, 2001); recombination could nevertheless be suppressed in the region. Furthermore, even in low-recombination regions, exchange may occur by gene conversion. Crossing over and gene conversion rates need not be strongly correlated (Langley et al, 2000; Jensen et al, 2002).
Consistent with the view that S-loci rarely recombine is the observation that the flanking regions of S-loci of some species differ greatly in sequences between alleles with different specificities (Coleman and Kao, 1992; Chung et al, 1995; Matton et al, 1995). There are, however, few comparisons between variability in the S-locus region and those of flanking regions of other genes in the same species. Thus, it is not yet known whether diversity in the S-locus region is unusual in the genome. Explicit tests for genetic exchange (recombination or gene conversion) are thus needed at S-loci.
Attempts to test for recombination in the gametophytic S-locus have produced varying conclusions. Clark and Kao (1991) did not detect intragenic recombination in S-allele sequences of four species of Solanaceae, using two tests based on clustering of polymorphic sites (Stephens, 1985; Sawyer, 1989), but their sample size was small. However, some intragenic recombination at the S-locus has been inferred for several species of Solanaceae. S-locus sequence diversity is higher than at S-linked loci (unpublished results in McCubbin and Kao, 1999; Li et al, 2000) and inconsistent evolutionary histories were observed for the 5′and 3′ regions of the S-locus in two sets of four closely related P. inflata S-alleles, suggesting recombination (Wang et al, 2001). Schierup et al (2001) used the informative sites test (Worobey, 2001) and r2 test of recombination, and also found evidence for recombination in two species of Solanaceae, but not in P. inflata.
To test whether intragenic recombination is a general feature of the gametophytic S-locus, we here use the relationship between linkage disequilibrium and distance between variable sites (Awadalla and Charlesworth, 1999) to test for recombination in S-loci of 21 species of Solanaceae, Rosaceae and Scrophulariacae.
Methods
We obtained data from 21 species for which five or more cDNA S-allele sequences, more than 170 bp long were available. Most are partial sequences between conserved regions C2 and C5 (see Richman et al, 1996; Richman and Kohn, 2000; Vieira and Charlesworth, 2002). For each species, we combined the cDNA sequences with amino-acid sequences from exons deduced from genomic S-RNase gene sequences, where available (see Table 2). The amino-acid sequences were aligned using ClustalX v. 1.64b (Thompson et al, 1997). There are some alignment gaps, mostly in the hypervariable regions. Balancing selection acting on S-alleles should ensure that there is little differentiation between populations (Schierup et al, 2000), so allele samples sampled from the species as a whole, as here, are suitable for testing recombination.
S-alleles are under balancing selection, so the infinite sites model, which underlies most available methods for testing for or estimating recombination in DNA sequence data, is violated (see discussion in Awadalla and Charlesworth, 1999). The aligned amino-acid sequences were therefore tested for a relationship between measures of linkage disequilibrium and nucleotide distances between variable sites, using Spearman's rank correlation. Linkage disequilibrium measures depend on the variant frequencies at the sites compared (Lewontin, 1988; McVean, 2001). We therefore used both D′, which corrects for variant frequencies (Devlin and Risch, 1995; Jorde and Bamshad, 2000), and r2 values. The D′ and r2 values were calculated using DnaSP software (Rozas and Rozas, 1999). To obtain P-values, 1000 data sets were generated with the D′ and r2 values obtained, but with randomized distances between sites (Awadalla and Charlesworth, 1999). Sequential Bonferroni correction for multiple nonindependent comparisons was applied (Rice, 1989) to each type of test (see below).
Gene conversion or crossing over both lead to a decline of linkage disequilibrium with distance, provided that the length of conversion tracts are similar to the size of the region examined (Takahata and Satta, 1998a, 1998b; Wiehe et al, 2000). In our data sets, most sites are less than 700 bp apart. Although the average length of a typical plant gene conversion tract is not known, it is probably often less than this (Dooner and Martinez-Férez, 1997; Drouin et al, 1999; Fu et al, 2002). In Brassica S-loci, linkage disequilibrium was found to decay within 400 nucleotides (Awadalla and Charlesworth, 1999).
We did four analyses for species of Solanaceae, and three of them for the Rosaceae and Scrophulariaceae, whose introns lengths differ too much for the fourth analysis (see below). The first analysis (column labelled A in Table 1) used all nonsingleton polymorphic sites with two variants, excluding alignment gaps. Since selection might lead to concordant polymorphic amino-acid variants in functionally different alleles, which could mimic recombination (Sawyer, 1989), we also tested using third codon positions only, using all pairs of nonsingleton sites (column B in Table 1).
Introns are known in gametophytic S-allele sequences of several species (reviewed in Vieira and Charlesworth, 2002). All S-allele genomic sequences so far obtained from species of Solanaceae (N=14), Rosaceae (N=18) and Scrophulariaceae (N=36) have one intron in the HVa region, and in the genomic sequences from Scrophulariaceae the intron lengths vary (Vieira and Charlesworth, 2002). Five of the 18 genomic S-allele sequences from Rosaceae have a second intron at the cleavage site between the signal peptide and the C1 region (Ma and Oliveira, 2000). In Rosaceae and Scrophulariaceae, the distances between pairs of polymorphic sites that are separated by introns therefore differ between different pairs of alleles in a species. Linkage disequilibrium should still decay with distance, but the relationship with distance may be obscured by the uncertainty of the distances, that is, will be weaker than if we knew the true distances. Our tests are therefore conservative as they reduce the chance of detecting recombination. We therefore did a third test using only pairs of polymorphic sites that are not separated by introns in any of the sequences compared (column C in Table 1). For Solanaceae, the 13 introns that have been described are of similar sizes (ranging from 87 to 125 bp, average 103.62; the error of the mean is 3.62). For sequences from this family, we also performed an additional test by adding the average size of the intron to the cDNA distances between sites that are separated by an intron (column D in Table 1).
Where possible, the analyses were also repeated using data sets excluding highly diverged sequences. Pairwise Ks values were estimated by Nei and Gojobori's (1986) measure with Jukes–Cantor correction, which is suitable for highly variable sequences. Sets of sequences were then formed in which five or more sequences remain after excluding all pairs with Ks>0.45. This analysis could not be carried out for P. hybrida, L. peruvianum, L. andersoni, S. carolinense, S. chacoense, N. alata, or any of the Antirrhinum species because all sequences were highly diverged. Two of the nine species for which sets could be formed had two suitable nonoverlapping sets of sequences (W. maculata and P. longifolia).
Results and discussion
We found significant negative correlations for both D′ and r2 for a number of species. There was no evidence for recombination in the data from Antirrhinum species (Scrophulariaceae). Although the correlations are very small, three from the Solanaceae and Rosaceae are significant after sequential Bonferroni correction (W. maculata, L. andersonii and Malus × domestica; Table 1, part I). None of the species gave significant negative correlations for both D′ and r2 with all the different analyses applied, but L. andersonii gave significant negative correlations for both measures with three of them (Table 1, part I).
These conclusions differ from those of Schierup et al (2001), who found no evidence for recombination in L. andersonii by either method used, while the r2 test suggested recombination for P. crassifolia and S. carolinense. There are several possible reasons for the differences. First, Schierup et al (2001) exclude segregating sites at frequencies below 30%. For S. carolinense, when the Schierup et al (2001) data set is used, our approach detects no significant correlations between either D′ or r2 and distance, while Schierup et al (2001) found weakly significant correlations. Second, for L. andersonii, Schierup et al (2001) analyzed more alleles (22, while our data set had 11), but a two-fold smaller region of the S-locus. Distances between the segregating sites compared were thus much shorter than in our data and the number of data points used in the correlations is 7.8 times smaller. Applying our methods to the data set of Schierup et al (2001), D′ declines significantly with distance (P<0.001). Third, in the data set of Schierup et al (2001), all pairs of segregating sites were less than 150 bp apart, so it is surprising that recombination was detected by them but not by us, although clearly larger numbers of sequences make it more likely that clear patterns will be detected, provided that the length of sequence is sufficient. Applying our methods to the P. crassifolia data set of Schierup et al (2001), a significant correlation between r2 and distance is observed (data not shown). Finally, different degrees of coadaptation between different amino-acid sites may cause differences between the two studies. If coadaptation is primarily between amino acids in different parts of the molecule, linkage disequilibrium could extend across considerable distances, and a decline with distance would be undetectable unless only closely segregating sites are analysed.
For P. inflata, both we and Schierup et al (2001) found only weak evidence for recombination, in disagreement with the results of Wang et al (2001). Highly divergent sequences in our data set, and that of Schierup et al (2001) might, however, obscure evidence for recombination. Evidence for exchange in the distant past could have been obliterated by subsequent mutations (Clark, 1993) and, since most S-alleles are old, the same mutation could have occurred twice at the same site. It is also possible that recombination rarely happens between very dissimilar S-allele sequences. Wang et al's (2001) approach of removing the most divergent sequences from the data sets could thus be preferable for testing for recombination. Although some known variants are omitted, there is no reason to think that this would falsely produce the appearance of recombination. Part II of Table 1 shows results of analyses of the data sets in which five or more sequences remain after excluding highly diverged sequences (see Methods). Negative correlations significant after Bonferroni correction were found for both D′ and r2 for three data sets (P. inflata, P. dulcis and Malus × domestica). P. inflata gave significant negative correlations for both D′ and r2 with all four different ways of analyzing the data (columns A, B, C and D in Table 1, part II), and P. dulcis with two of the three methods used for this species. Two other species yielded nonsignificant test results, whereas their sequences suggested recombination when all sequences were included. These were the two subsets of W. maculata and P. longifolia sequences (W. maculata 1, W. maculata 2, and P. longifolia 1 and P. longifolia 2, in Table 1, part II). The difference may be due to the small size of these data sets, with consequent low power to detect recombination.
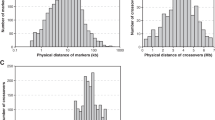
Data sets that produced significant negative correlations of both D′ and r2 with true or estimated genomic distance (columns C and D in Table 1, respectively) are illustrated in Figure 1a and b, respectively. L. andersonii and W. maculata show marked decreases of r2 with distance, in the analysis using all sequences. For P. inflata and P. dulcis, our analysis suggests recombination only when the most highly divergent sequences are excluded. Wang et al's (2001) analyses used four of the five S-allele sequences included in our analysis, so the agreement with their conclusion is expected.

Relationship between linkage disequilibrium, measured by r2, and the physical distance, measured in number of base pairs, using the data sets that showed significant results in which true (a) and estimated genomic distances (b) are known.
Our tests use related species, so that they are not independent, given that S-alleles may be maintained for very long evolutionary times. An S-allele from one species may therefore be more closely related to an S-allele from another species, or even from a different genus, than to another S-allele from the same species (sometimes called trans-specific evolution; Clark, 1993). Recombination events in an ancestor could therefore be detectable in more than one descendant species. Different results obtained for related species (eg P. avium and P. dulcis) may be due to true differences, or to low power to detect recombination in some data sets. Despite some inconsistent test results (perhaps not surprising, given the small sample sizes and sequence lengths available, and the well-known difficulties of detecting linkage disequilibrium as illustrated above), signs of genetic exchange are repeatedly found, and therefore seem difficult to ignore.
Although we cannot estimate the recombination frequency for gametophytic S-loci, the high level of silent site differences between S-alleles suggests that such recombination is rare. It is also not yet clear whether similar sequences experience much higher recombination rates than highly divergent ones. Nevertheless, even rare recombination could be an important factor in the evolution of these loci (Schierup et al, 2001), and in addition to mutation, could potentially generate new specificities (Wang et al, 2001).
Accession codes
Accessions
GenBank/EMBL/DDBJ
References
Awadalla P, Charlesworth D (1999). Recombination and selection at Brassica self-incompatibility loci. Genetics 152: 413–425.
Bernatzky R (1993). Genetic mapping and protein product diversity of the self-incompatibility locus in the wild tomato (Lycopersicon peruvianum). Biochem Genet 31: 369–373.
Charlesworth D, Charlesworth B (1998). Sequence variation: looking for effects genetic linkage. Curr Biol 8: R658–R661.
Chung IK, Lee SY, Ito T, Tanaka H, Nam HG, Takagi M (1995). The 5′ flanking sequences of two S alleles in Lycopersicon peruvianum are highly heterologous but contain short blocks of homologous sequences. Plant Cell Physiol 36: 1621–1627.
Clark AG (1993). Evolutionary inferences from molecular characterization of self-incompatibility alleles. In: Takahata N, Clark AG (eds) Mechanisms of Molecular Evolution, Sinauer Associates: Sunderland. pp 79–108.
Clark AG (1996). Population genetic aspects of gametophytic self-incompatibility. Plant Species Biol 11: 13–21.
Clark AG, Kao T-H (1991). Excess nonsynonymous substitution of shared polymorphic sites among self-incompatibility alleles of Solanaceae. Proc Natl Acad Sci USA 88: 9823–9827.
Coleman CE, Kao T-H (1992). The flanking regions of two Petunia inflata S alleles are heterogeneous and contain repetitive sequences. Plant Mol Biol 18: 725–737.
de Nettancourt D (1977). Incompatibility in Angiosperms. Springer-Verlag: Berlin.
Devlin B, Risch N (1995). A comparison of linkage disequilibrium measures for fine-scale mapping. Genomics 29: 311–322.
Dooner HK, Martinez-Férez IM (1997). Recombination occurs uniformly in the bronze gene, a recombination hotspot in the maize genome. Plant Cell 9: 1633–1646.
Drouin G, Prat F, Ell M, Clarke GD (1999). Detecting and characterizing gene conversions between multigene family members. Mol Biol Evol 16: 1369–1390.
Entani T, Iwano M, Shiba H, Takayama S, Fukui K, Isogai A (1990). Centromeric localization of an S-RNase gene in Petunia hybrida Vilm. Theor Appl Genet 99: 391–397.
Fu H, Zheng Z, Dooner HK (2002). Recombination rates between adjacent genic and retrotransposon regions in maize vary by 2 orders of magnitude. Proc Natl Acad Sci USA 99: 1082–1087.
Jensen MA, Charlesworth B, Kreitman M (2002). Patterns of genetic variation at a chromosome 4 locus of Drosophila melanogaster and D. simulans. Genetics 160: 493–507.
Jorde LB, Bamshad M (2000). Questioning evidence for recombination in human mitochondrial DNA. Science 288: 1931a
Langley CH, Lazzaro BP, Phillips W, Heikkinen E, Braverman JM (2000). Linkage disequilibria and the site frequency spectra in the su(s) and su(w(a)) regions of the Drosophila melanogaster X chromosome. Genetics 156: 1837–1852
Lewontin RC (1988). On measures of gametic disequilibrium. Genetics 120: 849–852
Li J-H, Nass N, Kusaba M, Dodds PN, Treloar N, Clarke AE et al (2000). A genetic map of the Nicotiana alata S locus that includes three pollen-expressed genes. Theor Appl Genet 100: 956–964.
Ma RC, Oliveira MM (2000). The RNase PD2 gene of almond (Prunus dulcis) represents an evolutionarily distinct class of S-like RNase genes. Mol Gen Genet 263: 925–933.
Matton DP, Mau SL, Okamoto S, Clarke AE, Newbigin E (1995). The S-locus of Nicotiana alata: genomic organization and sequence analysis of two S-RNase alleles. Plant Mol Biol 28: 847–858.
McCubbin AG, Kao T (1999). The emerging complexity of self- incompatibility (S-) loci. Sex Plant Reprod 12: 1–5.
McVean GAT (2001). What do patterns of genetic variability reveal about mitochondrial recombination? Heredity 87: 613–620.
Nei M, Gojobori T (1986). Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol 3: 418–426.
Nordborg M, Charlesworth B, Charlesworth D (1996). Increased levels of polymorphism surrounding selectively maintained sites in highly selfing species. Proc R Soc B 163: 1033–1039.
Rice WR (1989). Analyzing tables of statistical tests. Evolution 43: 223–225.
Richman AD, Uyenoyama MK, Kohn JR (1996). Allelic diversity and gene genealogy at the self-incompatibility locus in the Solanaceae. Science 273: 1212–1216.
Richman AD, Kohn JR (2000). Evolutionary genetics of self-incompatibility in the Solanaceae. Plant Mol Biol 42: 169–179.
Rozas J, Rozas R (1999). DnaSP version 3: an integrated program for molecular population genetics and molecular evolution analysis. Bioinformatics 15: 174–175.
Sawyer S (1989). Statistical tests for detecting gene conversion. Mol Biol Evol 6: 526–538.
Schierup MH, Vekemans X, Charlesworth D (2000). The effect of subdivision on variation at multi-allelic loci under balancing selection. Genet Res 76: 51–62.
Schierup MH, Mikkelsen AM, Hein J (2001). Recombination, balancing selection and phylogenies in MHC and self-incompatibility genes. Genetics 159: 1833–1844.
Strobeck C (1972). Heterozygosity in pin-thrum plants or with partial sex linkage. Genetics 72: 667–678.
Stephens JC (1985). Statistical methods of DNA sequence analysis–detection of intragenic recombination or gene conversion. Mol Biol Evol 2: 539–556.
Takahata N, Satta Y (1998a). Selection, convergence, and intragenic recombination in HLA diversity. Genetica 102–103: 157–169.
Takahata N, Satta Y (1998b). Footprints of intragenic recombination at HLA loci. Immunogenetics 47: 430–441.
ten Hoopen R, Harbord RM, Maes T, Nanninga N, Robbins TP (1998). The self-incompatibility (S) locus in Petunia hybrida is located on chromosome III in a region syntenic for the Solanaceae. Plant J 16: 729–734.
Thompson J, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997). The ClustalX window interface: flexible stategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25: 4876–4882.
Ushijima K, Sassa H, Tamura M, Kusaba M, Tao R, Gradziel TM et al. (2001). Characterization of the S-locus region of almond (Prunus dulcis). Analysis of a somaclonal mutant and a cosmid contig for an S haplotype. Genetics 158: 379–386.
Vekemans X, and Slatkin (1994). Gene and allelic genealogies at a gametophytic self-incompatibility locus. Genetics 137: 1157–1165.
Vieira CP, Charlesworth D (2002). Molecular variation at the self-incompatibility locus in natural populations of the genera Antirrhinum and Misopates. Heredity 88: 172–181.
Wang X, Hughes AL, Tsukamoto T, Ando T, Kao T (2001). Evidence that intragenic recombination contributes to allelic diversity of the S-RNase gene at the self-incompatibility (S) locus in Petunia inflata. Plant Physiol 125: 1012–1102
Wiehe T, Mountain J, Parham P, Slatkin M (2000). Distinguishing recombination and intragenic gene conversion by linkage disequilibrium patterns. Genet Res 75: 61–73.
Worobey M (2001). A novel approach to detecting and measuring recombination: new insights into evolution in virus, bacteria and mitochondria. Mol Biol Evol 18: 1425–1434.
Acknowledgements
CP Vieira is supported by the Fundação para a Ciencia e Tecnologia (SFRH/BPD/5592/2001).
Author information
Authors and Affiliations
Corresponding author
Appendix
Appendix
Rights and permissions
About this article
Cite this article
Vieira, C., Charlesworth, D. & Vieira, J. Evidence for rare recombination at the gametophytic self-incompatibility locus. Heredity 91, 262–267 (2003). https://doi.org/10.1038/sj.hdy.6800326
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.hdy.6800326
Keywords
This article is cited by
-
Large Scale Analyses and Visualization of Adaptive Amino Acid Changes Projects
Interdisciplinary Sciences: Computational Life Sciences (2018)
-
Overcoming self-incompatibility in grasses: a pathway to hybrid breeding
Theoretical and Applied Genetics (2016)
-
Identification, Evolutionary Patterns and Intragenic Recombination of the Gametophytic Self Incompatibility Pollen Gene (SFB) in Tunisian Prunus Species (Rosaceae)
Plant Molecular Biology Reporter (2016)
-
Gene duplication and genetic exchange drive the evolution of S-RNase-based self-incompatibility in Petunia
Nature Plants (2015)
-
Positional cloning of the s haplotype determining the floral and incompatibility phenotype of the long-styled morph of distylous Turnera subulata
Molecular Genetics and Genomics (2011)
