
Abstract
A study using allozymes and PCR fingerprinting was conducted to estimate the genetic diversity of Italian populations of two economically important cultivated fungal taxa, Pleurotus eryngii and P. ferulae. Very little is known about the genetic diversity distribution pattern of these taxa. Heterozygote deficiency was observed at few loci; in fact the inbreeding coefficients were not high, which demonstrates that mechanisms restrain the inbreeding act at the local level. Estimates of genetic differentiation indicated a pattern of greater variation within, rather than between, populations. These results were supported by AMOVA analysis, which attributed a low proportion of the total genetic variation to large geographical scale divergence, and indicated that most of the genetic diversity was because of differences within populations. This distribution pattern of genetic variation of P. eryngii and P. ferulae populations seems to be the result of high gene flow, by efficient basidiospore dispersal, and outcrossing mechanisms, which restrain inbreeding within populations.
Similar content being viewed by others

Introduction
Understanding the patterns of genetic diversity of species is fundamental not only for addressing questions concerning evolutionary processes and the development of conservation strategies, but also as a prerequisite for the efficient use of genetic resources in breeding programs. The interest in the genetic structure of natural populations has increased in the last few years owing to the necessity to broaden the knowledge of genetic variation in cultivated species. New approaches to the study of genetic variation from wild species to cultivated varieties, mediated by information on molecular markers, are promising avenues to exploit wild genetic resources. With fungi, breeding programs can be hindered by the lack of data on the extent of genetic variation in the natural populations. In fact, despite the economic importance of species in general or fungi in particular, little was known until recently about their natural history and evolutionary biology, for example, the evolutionary forces shaping natural populations and the available genetic variability.
The 11 species of the genus Pleurotus (Basidiomycota) are among the more conspicuous fungi causing wood decay in terrestrial ecosystems worldwide, and are widely collected and cultivated as edible fungi (Chang, 1993; Chang, 1996). They exhibit bipolar heterothallism; single-spore cultures from individual fruit bodies are of two kinds with several mating types in the population that efficiently promote outbreeding. The interaction of compatible monokaryons, derived from single basidiospores, results in a dikaryon (Elliot, 1994). There are few investigations on the population genetics of these species, and these only concern cultivated species, such as P. ostreatus (Jacq.: Fr.) P. Kumm.
The P. eryngii (DC.: Fr.)Quèl complex is distributed in the temperate Mediterranean region, in South Germany, Hungary, Slovakia and in Central Asia. Four varieties belong to this complex, two of which have recently been classified as separate species: P. eryngii and P. ferulae Authority (Urbanelli et al, 2002) The two taxa, morphologically similar in the juvenile phase, can be discriminated at maturity by the shape, color and diameter of the pileus and the size of the basidiospore (Hilber, 1982). They colonize the roots and the stems of some Apiaceae: P. eryngii that prevalently grows on the roots of Eryngium campestre, distributed throughout Western Europe, and P. ferulae grows mostly on the roots of Ferula communis, distributed throughout the Mediterranean region. These two host plants occur widely in Italy. Both mushroom taxa have been described as weak parasitic entities of these plants, although according to some authors they exhibit certain differences: P. eryngii is mainly saprophytic, while P. ferulae is mostly pathogenic (Cailleux et al, 1981; Urbanelli et al, 2002). Recent studies conducted on the genetic differentiation and the reproductive isolation of P. eryngii and P. ferulae populations have revealed that they are two distinct, genetically and reproductively isolated, entities (Urbanelli et al, 2002). The identification of molecular markers has allowed a clear discrimination of the two taxa, and the analysis of sympatric populations. The industrial production of these two taxa, under one name, P. eryngii, is well-developed in Italy and is currently under way in Europe. Hence, the need for a thorough knowledge of the genetic variation within and among populations of the two species.
In this paper, we report data on the use of informative markers systems to quantify the genetic diversity and understand the structure of natural Italian populations of P. eryngii and P. ferulae. We make inferences on the mating system and other factors affecting the observed levels and patterns of genetic variation.
Materials and methods
Studied samples and their geographic origin
We collected 123 wild fruitbodies of P. eryngii, near host E. campestre plants in six Italian localities and 261 wild fruitbodies of P. ferulae near Ferula communis in 13 Italian localities (). According to the sampling method, different fruitbodies might be derived from the same mycelium. To avoid the consequences of including the same individual several times, we collected each individual fungus from near different plants and at least 1 or 2 m apart.
The samples were morphologically identified by shape, color and diameter of pileus and size of the basidiospore.
Fruitbodies enzymatic extraction
Once in laboratory, each specimen was cut into small pieces and frozen in Eppendorf tubes at −80°C for later electrophoresis. Single pieces of fruitbodies were ground and dissolved in an extraction buffer (20 mM Tris–HCl at pH 7, 2 mM EDTA, 14 mM β-mercaptoethanol, 10 mM MgSO4, 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride and 0.1 M sorbitol) modified from Chalot et al (1989).
Allozyme gel electrophoresis
Homogenates were absorbed into 3 × 5 mm pieces of chromatography paper (Whatman 3MM) and placed on 10% starch gels. Standard horizontal electrophoresis was carried out at 7–8 V/cm for 4–6 h at 5°C. We analyzed up to 14 enzyme systems for each sample, encoded by 18 putative loci: α-Glycerophosphate dehydrogenase (α-Gpdh, EC 1.1.1.8), lactate dehydrogenase (Ldh, EC 1.1.1.28), malate dehydrogenase (Mdh-1, Mdh-2, EC 1.1.1.37), isocitrate dehydrogenase (Idh-1, Idh-2, EC 1.1.1.42), NADH Diaphorase (Dia, EC 1.6.2.2), superoxide dismutase (Sod-1, EC 1.15.1.1), aspartate aminotransferase (Aat-1, Aat-2, EC 2.6.1.1), phosphoglucomutase (Pgm, EC 5.4.2.2), leucine aminopeptidase (Lap, EC 3.4.11), peptidase (Leu–Ala) (Ap-1, Ap-2, EC 3.4.11), peptidase (Leu–Leu) (Pep-2, Pep-3, EC 3.4.11), Mannose phosphate isomerase (Mpi, EC 3.4.11), glucose phosphate isomerase (Gpi, EC 5.3.1.9). These loci included ≈50% of fast- and 50% of slow-evolving loci, sensu, Koehn and Eanes (1978). The electrophoretic procedures are reported in detail in Urbanelli et al (2002).
Allozymes were enumerated according to their decreasing mobility (from the most anodal) relative to the most common allele (=100) in a reference population (Monte Romano, Latium).
PCR-fingerprinting gel electrophoresis
Total genomic DNA was extracted from 15 mg of mycelia according to Noel and Labarére (1987). The primer used for PCR amplification was the internal repetitive sequence of bacteriophage M13 DNA (5′-GAGGGTGGCGGTTCT-3′) (Latouche et al, 1997). The band pattern that originated after 2 h (90 V) agarose gel (1.2%) electrophoresis was analyzed by the Gel-Compare 3.1 software (Applied Maths, Kortrijk, Belgium).
Allozyme data analysis
Allele frequencies were calculated for each population. Genotypes were computed for each individual in a population and used to estimate genetic parameters.
Genotype frequencies were tested for departures from Hardy–Weinberg equilibrium expectations with the exact probability test implemented by BIOSYS-1 (Swofford and Selander, 1981). The extent of genetic variation in each population was quantified by determining the number of alleles per locus (A), the percentage of polymorphic loci (P), 95% criterion, and the unbiased heterozygosity (H). The samples were of similar size and so the bias in the parameters of genetic variability did not affect the conclusions. The index of inbreeding, defined as FIS=1−(H0/HE), was calculated for each population over all polymorphic loci, where H0 and HE correspond to average observed and expected heterozygosities, respectively. The probability of outcrossing t was estimated using the equilibrium value of FIS, which is defined as (1−t)/(1+t) or t=(1−FIS)/(1+FIS) (Hartl, 1987).
Gene diversity statistics were obtained using the method of Nei (1987). The total allelic diversity is represented by HT, which is partitioned into the mean allelic diversity within population, HS, and the allelic diversity among populations, DST. These quantities are related by the expression HT=HS+DST. The proportion of the total allelic diversity found among populations, GST, was calculated as the ratio DST/HT.
The gene flow among populations, or genetically effective migration rate (Nm), was estimated using Wright's FST (Wright, 1951). The effective migration rate, estimated by the fixation index FST, is defined with the formula Nm=(1/FST)−1/4 (Wright, 1969; Slatkin, 1987). This formula assumes the infinite-island model of population structure and gene flow; very few populations conform to this assumption, but it provides a useful approximation of the relative magnitude of gene flow.
PCR-fingerprinting data analysis
All individuals were scored for the presence or absence of fingerprinting fragments, and the data were entered into a binary data matrix as discrete variables (1=presence; 0=absence). Four populations of P. eryngii and four population of P. ferulae were studied ( Table 4). The same specimens analyzed using isozymes were used for PCR-fingerprinting analysis. To generate similarity matrices, the data were analyzed using Excoffier et al (1992) algorithm, with the software RAPD analysis, ver. 1.04 (Armstrong et al, 1993).
The AMOVA analysis was performed on the fingerprinting haplotypes, and the Euclidean distances were calculated between all pairs of haplotypes (Excoffier et al, 1992; Huff et al, 1993). This analysis was used to split the total genetic variation into that occurring within populations [ΦST=(σa2+σb2)/σ2)], among populations within regions (ΦSC=σa2/σ2) and between regions [ΦCT=σ2/(σb2+σc2)]. The associated test for the homogeneity of molecular variance was carried out. All the variance components were tested statistically by nonparametric randomization tests using 2000 permutations. These analyses were performed with the WINAMOVA program provided by Excoffier (University of Geneva, Switzerland).
Results
Allozyme analysis
Genetic polymorphism: The 18 putative enzyme loci investigated allowed us to study the genetic variation of six populations of P. eryngii and 13 populations of P. ferulae. Polymorphism was observed at 13 enzyme loci (Ldh, Mdh-1, Mdh-2, Idh-2, Dia, Aat-1, Aat-2, Pgm, Ap-2, Pep-2, Pep-3 Mpi and Gpi). Among these, four (Mdh-2, Dia, Pep-2 and Pgm) were diagnostic among taxa, allowing clear identification of P. eryngii and P. ferulae samples from the field ( Table 1). The results of Hardy–Weinberg tests showed that genotype frequencies at most polymorphic loci did not depart significantly from the expected values. Significant deviations (P<0.05), always associated with heterozygote deficiencies, were observed in the P. eryngii populations for the loci Ldh (population 1), Mdh-2 (2), Dia (6) and Mpi (3–6), and in the P. ferulae populations for the loci Ldh (9 and 10), Dia (13 and 14), Pgm (19), Mpi (7,11, 12, 14–16 and 18) and Pgi (18).Table 2
The genetic variability parameters were higher in P. eryngii than in P. ferulae samples (Table 2). The most variable P. eryngii population was Monte Romano (A=2.1, P95=0.72 and Ho=0.240); the least variable was Ceglie Messapico (A=1.4, P95=0.33 and Ho=0.115). Within P. ferulae the most and variable population was Castel del Monte (A=1.6, P95=0.44 and Ho=0.165); Macomer was the least variable (A=1.5, P95=0.39 and Ho=0.085).
The estimates of the inbreeding coefficient (FIS), averaged over all polymorphic loci in each population, were positive and only in one case were they significant. The FIS values were 0.235 in P. eryngii and 0.262 in P. ferulae, reflecting a low autogamy rate. The outcrossing rates, estimated from the FIS index averaged over all polymorphic loci, were high, ranging from 0.485 to 0.914 (mean=0.632) in P. eryngii and from 0.416 to 0.721 (mean=0.592) in P. ferulae ( Table 2).
Genetic diversity and gene flow: Averaged over all loci, the total genetic diversity HT was 0.233 for P. eryngii and 0.172 for P. ferulae taxa ( Table 3). The genetic diversity within populations was responsible for most of this diversity (HS=0.212 in P. eryngii and HS=0.165 in P. ferulae). In fact, the coefficients of genetic differentiation among populations, GST′=0.100 in P. eryngii and 0.045 in P. ferulae, showed that a large amount of genetic diversity lies within populations. Genetic differentiation between populations differed greatly among loci and were highest for Dia (GST′=0.241), Ap-2 (GST′=0.058), Mpi (GST′=0.123) in P. eryngii, and for Mpi (GST′=0.082), Idh-2 (GST′=0.072) and Pgm (GST′=0.080) in P. ferulae. However, the locus Pep-2 was fixed for an allele within P. ferulae populations. While the largest proportion of genetic diversity was because of variation within populations, HS=0.212 in P. eryngii and HS=0.165 in P. ferulae, the remaining variation, DST=0.021 and 0.007, was because of allele frequency differences between populations.
The gene flow (Nm) estimates obtained for each locus by the FST (NmFST) were in P. eryngii (NmFST=3.50) and in P. ferulae (NmFST=5.43). The different estimates of gene flow observed between the two taxa may be because of the lesser number of samples of P. eryngii compared to P. ferulae.
PCR-fingerprinting
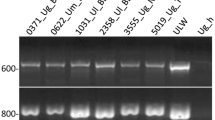
Genetic polymorphism: The minisatellite M13 amplified 2217 scored bands in the 81 specimens of Pleurotus eryngii (populations 2, 3, 5 and 6) and 87 specimens of P. ferulae (9, 12, 15 and 19). Of these, only well-amplified (sharp gel signal) and reproducible (in all experiments) bands were considered for the analysis (Figure 1). The number of bands obtained varied from 4 to 13 for each specimen. Three bands appeared to discriminate the two taxa: P. eryngii were characterized by a band at 1140 bp, while P. ferulae showed two bands at 300 and 1200 bp (Figure 1). The analysis of banding patterns revealed polymorphism both between and within populations of the two taxa. A similar number of specific bands (11–15) was found in P. eryngii and P. ferulae populations. The level of intrapopulational polymorphism varied from one population to another. None of the P. eryngii and P. ferulae populations were completely monomorphic; the polymorphic bands varied from 30 to 50% within P. eryngii and from 40 to 50% within P. ferulae populations ( Table 4). These differences were not significant between taxa.

PCR-fingerprinting of genomic DNA of P. eryngii and P. ferulae with minisatellite M13. Lanes 1, 16 100 bp molecular-weight marker. Lanes 2–8, 10, 14 P. eryngii; lanes 9,11–13, P. ferulae; lane 15 control without template DNA.
Genetic diversity: AMOVA for the two taxa indicated significant genetic differences (P<0.0005) within populations, but no significant difference was observed between regions ( Table 5). In P. eryngii the total genetic diversity was ascribed to individual differences within populations (61.3%), to population differences within regions (21.5%) and to regional divergence (17.2%). Most of the genetic variation observed within regions was because of differences within populations (65.4%), and the genetic variation between regions was accounted for by the variation within regions (72.8%). The total genetic diversity in P. ferulae was mostly because of variations within populations (84.1%), as compared with variation within regions (11.4%) and with regional divergence (4.5%). Furthermore, genetic variation within and between regions was ascribed to individual differences (86.2 and 88.8%, respectively). Both approaches, allozymes and PCR-fingerprinting, yield an overall similar trend; genetic diversity is greater within than among populations.
Discussion
The principal aim of this study was to document the level and distribution of genetic diversity in Italian populations of two Pleurotus species, P. eryngii and P. ferulae.
The level of genetic variation was evaluated by PCR-fingerprinting and allozyme analyses in these populations. With PCR-fingerprinting, the frequencies of polymorphic bands were not significantly different between P. eryngii and P. ferulae populations. The values of the genetic parameters with allozymes were lower (P=0.45, Ho=0.161 in P. eryngii and P=0.40, Ho=0.117 in P. ferulae) than the values observed in some basidiomycetes (eg in Mycena rosea, Ho=0.217, Boisselier-Dubayle et al, 1996; Agaricus campestris Ho=0.281, May and Rose, 1982). Similar values of genotypic diversity were recorded by Zervakis et al (1994) in Pleurotus species: P. ostreatus (0.093), P. pulmonarius (0.083) and for P. eryngii complex (0.137). The comparison with the above results is somehow problematic since the authors derived their values from either few isolates or few gene–enzyme systems or both, so they might not be strictly equivalent to our results. High levels of genetic variation are generally expected for wild organisms that reproduce sexually and have broad ecological niches and a wide geographical distribution (Babbel and Selander, 1974; Hamrick and Godt, 1990; James et al, 1999). By contrast, low levels of variation were observed in species with a restricted distribution, or that have long been cultivated for commercial purposes, or in populations with just a few individuals, or in species that reproduce exclusively asexually (Old et al, 1984; Burdon and Roelfs, 1985). For example, high levels of genetic variation were observed in broadly distributed species, such as Schizophyllum commune (James et al, 1999). Nevertheless, high genetic variation has been observed in species such as Mycena rosea, with narrow distributions (only in Western Europe) (Boisselier-Dubayle et al, 1996). High genetic variability would therefore seem attributable to species of wild origin, which reproduce sexually. Pleurotus eryngii and P. ferulae have both these characteristics. They are distributed throughout rather circumscribed areas and are related to specific host plants, E. campestre and F. communis, respectively, which could lead to the values of genetic variability observed within populations of the two species. Our results could fit with Chen et al (1992) hypothesis, which ascribed the moderate level of genetic variation in the fungi of the genus Pythium to selection pressures consistent with host-specific evolutionary development. This hypothesis will remain as such until a deeper knowledge of the degree of genetic variation in fungi is acquired.
Heterozygote deficiency within some populations of the two species was found at few loci tested. Since inbreeding is revealed through heterozygote deficit, it seems to have little influence on the populations of the two taxa. In fact the inbreeding levels observed were not high (FIS=0.235 in P. eryngii and 0.262 in P. ferulae populations). High inbreeding coefficients usually range between 0.80 and 1.00, and are found in species where inbreeding prevails. Conversely, outcrossing values were higher than inbreeding coefficients, and ranged from 0.485 to 0.914 in P. eryngii and from 0.416 to 0.721 in P. ferulae. This would demonstrate that mechanisms that restrain inbreeding are acting at the local level, in agreement with Bresinsky et al (1987) results on the genus Pleurotus. With crossbreeding experiments they demonstrated that both inbreeding and outcrossing are possible; however, outcrossing can be favored by the multiple allelism of the mating factors A and B.
The estimates of genetic differentiation, averaged over all loci, indicated that the total genetic diversity was low (GST′=0.100 in P. eryngii and GST′=0.045 in P. ferulae), and a high level of variation was found within populations (HS=0.212 in P. eryngii and HS=0.165 in P. ferulae). Similar results were obtained by AMOVA analysis of the populations, which demonstrated that a great portion of the total genetic variation could be attributed to the differences within populations, revealing low genetic differentiation over moderate (within regions) and large geographic distances (among regions).
Partitioning of genetic variation within, rather than among, populations has been observed also in other Basidiomycete species. Mating system and gene flow are some of the most important factors that determine genetic structure. Some studies have led to the generalization that outcrossing species maintain most genetic variability within, rather than among, populations, whereas inbreeding species partition most variability among populations (James et al, 1999). The gene flow, by spore dispersal, appears to be another major force in the determination of the local genetic structure of populations. Several studies on lignicolous, humicolous and saprotrophic fungi have shown that, when the local populations are maintained chiefly by basidiospore dispersal, several genetically different individuals occupy a discrete zone within a substrate: Pleurotus ostreatus (Kay and Vilgalys, 1992); Mycena rosea (Boisselier-Dubayle et al, 1996); Collybia subnuda and Marasmiellus praecutus (Murphy and Miller, 1993). In contrast, the populations maintained by basidiospore dispersal and subsequent rapid vegetative spread on nearby substrates consist of few individuals or just one individual with genetically identical clones: Armillaria bulbosa (Smith et al, 1992); Lycoperdon pyriforme (Huff, 1993). In this case the transition to a new substrate may occur not by the arrival of spores directly to the substrate, but from colonies initially established in the leaf litter or soil. These two different dispersal processes in fungal populations could reflect the different genetic structure of the populations: low genetic diversity and high gene flow between populations were observed in those populations that are primarily maintained through basidiospore dispersal.
The data we present show a similar genetic diversity partitioning pattern for P. eryngii and P. ferulae. Most of the observed variation is contained within local populations, which is likely to be the result of high gene flow, by efficient patterns of dispersal in sexual, outcrossing species. The high values of gene flow observed in P. eryngii (NmFST=3.50) and P. ferulae (NmFST=5.43), and the outcrossing mechanisms that restrain inbreeding seem to play an important role in the partitioning of genetic diversity of the Italian populations of P. eryngii and P. ferulae. Our findings agree with the results obtained by Boisselier-Dubayle et al (1996) for the saprophytic fungus Mycena rosea and would indicate basidiospore dispersal as a primary mechanism for dykarion establishment in the two species. Further research into the spatial distribution and genetic relationships among individuals will be necessary to confirm our conclusions.
References
Armstrong J, Gibbs A, Peakall R, Weiller G (1993). RAPDistance Programs; Version 1.04 for the analysis of patterns of RAPD fragments. Australian National University, Canberra, Australia.
Babbel GR, Selander RK (1974). Genetic variability in edaphically restricted and widespread plant species. Evolution 28: 619–630.
Boisselier-Dubayle MC, Perreau-Bertrand J, Lambourdiere J (1996). Genetic variation in wild populations of Mycena rosea. Mycol Res 100: 753–758.
Bresinsky A, Fisher M, Meixner B, Paulus W (1987). Speciation in Pleurotus. Mycologia 79: 234–245.
Burdon JJ, Roelfs AP (1985). The effect of sexual and asexual reproduction on the isozyme structure of population Puccinia graminis. Phytopathology 75: 1068–1073.
Cailleux R, Diop A, Joly P (1981). Relations d'interfertilitè entre quelques reprèsententes des Pleurotes des Ombrellifères. Bull Soc Mycol Fr 97: 97–124.
Chalot M, Brum A, Botton B (1989). Occurrence and distribution of the fungal NADP-dependent glutamate dehydrogenase in spruce and beech ectomycorhizas. In: Gustav FWD, D. Werner, Müller P (eds) Fast Growing Trees and Nitrogen Fixing Trees. Stuttgart. pp 324–327.
Chang ST (1993). Mushroom biology: the impact on mushroom production and mushroom products. In: Chang ST, Busweell JA, Chiu SW et al (eds) Mushroom Biology and Mushroom Products. Chinese University Press: Hong Kong. pp 3–20.
Chang R (1996). Functional properties of edible mushrooms. Nutr Abstr Rev 54: 91–93.
Chen W, Schneider RW, Hoy JW (1992). Taxonomic and phylogenetic analyses of ten Pythium species using isozyme polymorphisms. Phytopathology 82: 1234–1243.
Elliot CG (1994). Reproduction in fungi. In: Genetical and Physiological Aspects, Chapman and Hall: London.
Excoffier L, Smouse PE, Quattro JM (1992). Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131: 479–491.
Hamrick JL, Godt MJ (1990). Allozyme diversity in plant species. In: Brown AHD, Clegg MT, Kahler AL, Weir BS, Plant population Genetics, Breeding and Genetic Resources, Sinauer: Sunderland, MA. pp 43–63.
Hartl DL (1987). A Primer of Populations Genetics. Sinauer Associates Inc.: Sunderland, MA.
Hilber O (1982). Die Gattung Pleurotus (Fr.) Kummer unter besonderer Bercksichtigung des Pleurotus eryngii-Formenkomplexes. Bibliotheca Mycologica 87: 448.
Huff DR, Peakall R, Smouse PE (1993). RAPD variation within and among natural populations of outcrossing Buffalo grass (Buchloe dactyloides (Nutt. Engelm.) Theor Appl Genet 86: 927–934.
James TY, porter D, Hamrick JL, Vilgalys R (1999). Evidence for limited intercontinental gene flow in the cosmopolitan mushroom, Schizophyllum commune. Evolution 53: 1665–1667.
Kay E, Vilgalys R (1992). Spatial distribution and genetic relationships among individuals in a natural populations of the oyster mushroom Pleurotus ostreatus. Mycologia 84: 173–182.
Koehn RK, Eanes WF (1978). Molecular structure and protein variation within and among populations. Evol Biol 11: 39–100.
Latouche GN, Daniel HM, Lee OC, Mitchell TG, Sorre, TC, Myer W (1997). Comparison of use of phenotypic and genotypic characteristics for identification of species of the anamorph genus Candida and related teleomorph yeast species. J Clin Microbiol 35: 3171–3180.
May B, Rose DJ (1982). Genetic variation and joint segregation of biochemical loci in the common meadow mushroom Agaricus campestris. Biochem Genet 20: 1165–1173.
Murphy JF, Miller OK (1993). The population biology of two litter decomposing agarics on a southern Appalachian mountain. Mycologia 85: 769–776.
Nei M (1987). Molecular Evolutionary Genetics, Columbia University Press: New York.
Noel J, Labarére J (1987). Isolation of DNA from Agrocybe aegerita for the construction of a genomic library in E. coli. Mushroom Science 12: 187–201.
Old KM, Moran GF, Bell JC (1984). Isozyme variability among isolates of Phytophthora cinnamoni from Australia and Papua New Guinea. Can J Bot 62: 2016–2022.
Slatkin M (1987). Gene flow and the geographic structure of natural populations. Science 236: 787–792.
Smith ML, Bruhn JN, Anderson JB (1992). The fungus Armillaria bulbosa is among the largest and oldest living organisms. Nature 356: 428–431.
Swofford DL, Selander RR (1981). BIOSYS-1: a FORTRAN program for the comprehensive analysis of electophoretics data in population genetics and systematics. J Heredity 72: 281–283.
Urbanelli S, Fanelli C, Fabbri AA Della Rosa V, Maddau L, Marras F, Reverberi M (2002). Taxonomic studies on two taxa of the Pleurotus eryngii complex: Pleurotus eryngii var. eryngii and P. eryngii var. ferulae (Quel.:Fr.). Biol J Linnean Soc 75: 125–136.
Wright S (1951). The genetical structure of populations. Ann Eugen 15: 323–354.
Wright S (1969). Evolution and the Genetics of Populations, Vol 2. The theory of gene frequencies. Chicago University Press: Chicago.
Zervakis G, Sourdis J, Balis C (1994). Genetic variability and systematics of eleven Pleurotus species based on isoenzyme analysis. Mycol Res 98: 329–341.
Acknowledgements
We are grateful to two anonymous reviewers for very helpful comments on the manuscript; Prof. Nascetti, Dr L Nini, Dr A Ricelli, Dr V Tasca and Dr S Zjalic for their invaluable cooperation. The research was carried out with grants from the Ministero delle Protezioni Agricole and the Life Science Project of the European Community.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Urbanelli, S., Della Rosa, V., Fanelli, C. et al. Genetic diversity and population structure of the Italian fungi belonging to the taxa Pleurotus eryngii (DC.:Fr.) Quèl and P. ferulae (DC.:Fr.) Quèl. Heredity 90, 253–259 (2003). https://doi.org/10.1038/sj.hdy.6800239
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.hdy.6800239
Keywords
This article is cited by
-
The saprotrophic Pleurotus ostreatus species complex: late Eocene origin in East Asia, multiple dispersal, and complex speciation
IMA Fungus (2020)
-
Genetic diversity of Phytophthora palmivora isolates from Indonesia and Japan using rep-PCR and microsatellite markers
Journal of General Plant Pathology (2019)
-
Genetic diversity and population structure of Chinese Lentinula edodes revealed by InDel and SSR markers
Mycological Progress (2016)
-
AFLP based assessment of genetic relationships among shiitake (Lentinula ssp.) mushrooms
Molecular Biology Reports (2012)
-
Analysis of population structure of Puccinia striiformis in Yunnan Province of China by using AFLP
European Journal of Plant Pathology (2011)
