
Abstract
RT-PCR was used to obtain the first estimates from natural populations of allelic diversity at the RNase-based gametophytic self-incompatibility locus in the Rosaceae. A total of 20 alleles were retrieved from 20 Sorbus aucuparia individuals, whereas 17 alleles were found in 13 Crataegus monogyna samples. Estimates of population-level allele numbers fall within the range observed in the Solanaceae, the only other family with RNase-based incompatibility for which estimates are available. The nucleotide diversity of S-allele sequences was found to be much lower in the two Rosaceae species as compared with the Solanaceae. This was not due to a lower sequence divergence among most closely related alleles. Rather, it is the depth of the entire genealogy that differs markedly in the two families, with Rosaceae S-alleles exhibiting more recent apparent coalescence. We also investigated patterns of selection at the molecular level by comparing nucleotide diversity at synonymous and nonsynonymous sites. Stabilizing selection was inferred for the 5′ region of the molecule, while evidence of diversifying selection was present elsewhere.
Similar content being viewed by others


Introduction
Extreme polymorphism at the self-incompatibility loci of flowering plants has long attracted the attention of population biologists because the properties of these systems can be deduced from an understanding of their simple inheritance. This is particularly true of single-locus gametophytic self-incompatibility (GSI) in which a haploid pollen tube is rejected if it carries either allele found in the diploid pollen recipient. Under this simple system of self-recognition and rejection, alleles are subject to negative frequency-dependent selection because rare alleles have access to more mates while common alleles are more frequently rejected. The strength of diversifying selection becomes attenuated as the number of alleles in a population increases until a balance is reached between selection favouring the addition of alleles through migration or mutation and drift removing them (Wright, 1939). Finally, unless other forms of selection are acting to favour specific S-alleles, the expected frequency of each of n alleles in a population at equilibrium is 1/n (isoplethy).
The study of gametophytic self-incompatibility systems began by using diallele crossing designs to estimate the number of S-alleles in natural populations (reviewed in Lawrence, 2000). These studies established that large numbers of alleles were present in natural populations of eight GSI species, as predicted under frequency-dependent selection. More recently, advances in understanding the molecular basis of incompatibility systems in several plant families allowed the development of PCR based methods for the study of S-allele variation. PCR methods can identify S-alleles in taxa not amenable to greenhouse crossing and allow the study of S-allele sequence diversity as well as number. Key to the development of the research reported here was the finding that RNases are the stylar product of the gametophytic S-locus responsible for self-pollen recognition and rejection in three families of flowering plants, the Solanaceae, Scrophulariaceae, and Rosaceae (McClure et al, 1989; Sassa et al, 1996; Xue et al, 1996).
In conjunction with our ability to obtain sequences of S-alleles, advances in the application of coalescence theory to systems under balancing selection, one form of which is negative frequency-dependent selection, provide a framework for the comparative study of S-allele sequence diversity complementary to investigations of S-allele number. A key prediction of coalescence theory applied to systems under balancing selection is that balanced polymorphism should persist far longer than neutral polymorphism (Takahata, 1990; Vekemans and Slatkin, 1994). This is an additional outcome of frequency-dependent selection which tends to prevent rare alleles from drifting to extinction.
Several studies of S-allele number and sequence diversity in natural populations of Solanaceae have been published using PCR methodology (Richman et al, 1995, 1996a; Richman and Kohn, 1999; Richman, 2000; Lu, 2001; Wang et al, 2001). These have borne out the prediction of ancient polymorphism at the S-locus with the common observation of transpecific evolution (sister alleles from different species or genera). Transpecific evolution is also found in other systems under balancing selection such as the MHC loci of vertebrates (Edwards and Hedrick, 1998) and the mating type loci of fungi (Wu et al., 1998), and indicates that much current polymorphism predates the origin of extant taxa. Because of this greatly increased time depth, polymorphism at loci under balancing selection may allow a window into history at time-depths unapproachable using studies of neutral variation (Takahata, 1990).
Prior to this study no data on allele number or sequence diversity of S-RNases from natural populations of Rosaceae were available to compare with those from the Solanaceae. Analysis of several S-alleles from cultivated Rosaceae (apples, pears, almonds, and cherries) suggested strikingly lower levels of species-specific sequence diversity than in the Solanaceae (Ishimizu et al, 1998). This study had three goals: (1) to estimate S-allele number in natural populations of Crataegus monogyna and Sorbus aucuparia, two species of Rosaceae that have previously been reported to be self-incompatible (Grime et al, 1988; Raspé et al, 2000a); (2) to compare levels of S-allele sequence diversity, transpecific polymorphism, and overall coalescence depth, with those found in the Solanaceae; (3) to analyze patterns of selection at the molecular level in the two families. In the Solanaceae, evidence for positive selection has been obtained in several studies (Clark and Kao 1991; Richman et al, 1996b). Comparison of patterns of S-allele diversification in the Solanaceae and Rosaceae allows a rare chance to see how similar or different are two outcomes of the same evolutionary process in separate lineages.
Materials and methods
Plant material
Twigs bearing inflorescences were sampled in a single population of C. monogyna and four closely adjacent sites containing S. aucuparia. All sites were located on the Plateau des Tailles (Belgium) and S. aucuparia sampling locations were separated from each other by less than 5 km. Twigs were brought back to the laboratory and c. 20–30 (S. aucuparia) or 10–15 (C. monogyna) styles of freshly opened flowers or flower buds 1–2 days prior to anthesis were frozen in liquid nitrogen and stored at −80°C until further use.
S-allele amplification, cloning and sequencing
We used the RT-PCR methodology developed by Richman et al (1995) to amplify S-alleles. Styles were ground on dry ice and stylar RNA was isolated using TRIZOL™ (Gibco BRL) according to the manufacturer’s instructions, and resuspended in 20 μl of water. cDNA synthesis was performed with 2–3 μl of RNA solution, using the cDNA Cycle® kit (Invitrogen) or the Sensiscript™ RT kit (Qiagen), with similar results. A 2 μl aliquot of the cDNA synthesis reaction was used for amplification in a 35 μl reaction mix containing 1 × Taq buffer, 1 mM MgCl2, 200 μM of each of the four dNTPs, 50 ng of each primer and 0.5 U of Taq Polymerase. Reactions were covered with one drop of mineral oil and amplified using one step of 4 min at 94°C, 35 cycles of 45 sec at 94°C, 2 min at 45°C and 1 min at 72°C, followed by one step of 12 min at 72°C. The PCR yield was generally low, so that it was necessary to run the PCR product on a 2% agarose gel, take a plug out of the band, and use this DNA as a template for a second PCR.
Degenerate primers used for PCR amplification were designed from the conserved regions 1, 2, and 5 (C1, C2 and C5; Kheyr-Pour et al, 1990; Sassa et al, 1996) of published S-allele sequences of Malus domestica and Pyrus spp. retrieved from GenBank (Table 1). In some individuals, only one allele could be amplified with the C1–C5 primers. In such cases, the other allele was amplified using more internal primers, C2 in place of C1, and/or R2 or R3 in place of C5. R2 and R3 reverse primers are complementary to a sequence starting 15 codons upstream of C5. The two alleles of each individual were identified by RFLP, and cloned using the Invitrogen TA cloning kit. Different cloned alleles were identified by RFLP and sequenced.
Analysis of the progeny from a half-compatible cross
In a half-compatible cross, only the allele of the pollen donor that differs from either allele of the pollen recipient is transmitted to the progeny. As a result, only two progeny genotypes are expected. This provides an opportunity to demonstrate the association between sequence variants and the self-incompatibility reactions of individual plants. We used a progeny from a controlled cross performed prior to the present study (Raspé, 2001), which we predicted to be half compatible based on the sequences of the parents amplified in this study. Progeny S-alleles were amplified directly from genomic DNA (see Raspé et al, 2000b for the DNA isolation procedure). The forward primer used (sequence: 5′-CAGTTGRAAATWATTTGGCCG-3′) was designed so that the intron was not included in the amplified sequence. Amplification products from both parents and progeny were digested with AluI, which provided a different profile for each of the three sequences.
S-allele number and frequencies
The total number of alleles in the population (N) was estimated by the method of Paxman (1963). The 95% likelihood interval of this estimator was computed according to O’Donnell and Lawrence (1984). These estimators assume that all alleles occur at the same frequency, as expected in a population at equilibrium. The assumption of isoplethy was tested using the Mantel (1974) statistic (see Campbell and Lawrence, 1981a). Because the distribution of the test statistic is likely to depart from the chi-square distribution for the sample sizes considered, its significance was evaluated by Monte Carlo simulation (see Richman et al., 1996a). One thousand random samples of r genotypes were drawn with replacement from a pool of n alleles at equal frequency. Using the number of genotypes sampled in each species as r and the estimated allele number as n, Mantel’s statistic was computed for each sample and the observed value compared to the distribution of sample values.
Sequence analyses
To assess transpecific evolution, we constructed neighbour-joining and maximum parsimony phylogenies of alleles from S. aucuparia, C. monogyna and related cultivated species, all members of the Rosaceae subfamily Maloideae. The phylogenies were rooted with an S-allele from cherry, Prunus avium (Rosaceae: Amygdaloideae).
To compare levels of sequence variation in Rosaceae to those found in Solanaceae, the following analyses were limited to the 125 amino acid positions downstream of C2, because comparable sequence data between C1 and C2 were unavailable for natural populations of Solanaceae. Alleles for which less than 125 codons were available downstream of C2 (one allele from S. aucuparia) were deleted from the analysis. Mean pairwise nucleotide diversity (π) for all sites was computed under the Kimura 2-parameter model implemented in the Arlequin 2.000 software package (Schneider et al, 2000). Pairwise diversity is influenced both by the divergence of each allele from its most closely related sister allele and by the total divergence (depth) in the genealogy. In order to separate these effects we used the DNAdist and Kitsch programs of the PHYLIP 3.572 computer package (Felsenstein, 1995) to generate pairwise distances under the Kimura 2-parameter model and to produce neighbour-joining genealogies with contemporaneous tips. From these we computed average terminal branch lengths and the depths of the genealogies for both Rosaceae and Solanaceae. Pairwise differences in terminal branch length were tested with the nonparametric Wilcoxon two-sample test.
Patterns of selection at the molecular level were analysed using the SNAP program (Korber, 2000) which estimates average pairwise nucleotide diversity at synonymous (Ds) and nonsynonymous (Dn) sites using the method of Nei and Gojobori (1986). We first explored patterns of diversity at synonymous and nonsynonymous sites across the molecule using the codon by codon estimates from SNAP to see if different forms of selction might be acting in different regions. We then compared estimates of Dn and Ds for given regions by Z-tests, as suggested by Kumar et al (2001).
Results
Allele numbers and frequencies
In all, 20 alleles were found in 20 S. aucuparia individuals, whereas 17 alleles were retrieved from 13 C. monogyna samples. All sequences were deposited to GenBank (accession numbers AF504253 to AF504290). A sequence alignment is also available from the authors upon request. Population allele number estimates and 95% likelihood intervals were 24.4 (20–34) and 26.7 (18–50) for S. aucuparia and C. monogyna, respectively. For comparison with other studies, it is useful to summarise the thoroughness of sampling using the repeatability statistic

where m is the number of alleles examined (equal to 2r), and n is the number of different alleles found (Campbell and Lawrence, 1981b). R = 0.54 and 0.39 in S. aucuparia and C. monogyna, respectively. Allele frequencies did not deviate from isoplethy in S. aucuparia (χ2 = 16.9; P > 0.10), whereas they were significantly unequal in C. monogyna (χ2 = 11.3; P < 0.05) chiefly because two alleles were sampled four times, an unexpected result given the small sample (Figure 1). Because of this deviation from isoplethy, we also estimated allele number in C. monogyna using the E2 estimator of O’Donnell and Lawrence (1984), which does not assume uniform allele frequencies, and obtained nearly the same estimate of allele number (27).

Observed and expected frequencies of sampling putative S-alleles. Observed frequencies reflect the number of times an allele was recovered one, two, or more times in the sample (20 plants for Sorbus aucuparia; 13 for Crataegus monogyna). Expected frequencies were generated using equation 6 of Yokoyama and Hetherington (1982) assuming populations containing 24.4 (S. aucuparia) and 26.7 (C. monogyna) alleles at uniform expected frequencies and sample sizes equal to those of the real data. Significance of Mantel’s (1974) χ2 statistic was evaluated using 1000 Monte Carlo samples of the same number of plants as examined in each species.
Half-compatible cross
As expected under gametophytic self-incompatibility, the progeny from the cross 8.25(S5S12) × 8.2(S1S5) showed only two genotypes, S1S5 and S1S12, indicating that offspring received only the compatible paternal allele. The observed ratio between the two genotypes was very close to the expected ratio of 1:1, S1S5 and S1S12 being observed 16 and 14 times, respectively.
Sequence variation
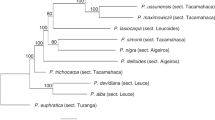
A neighbour-joining tree of alleles from S. aucuparia, C. monogyna, and previously published sequences from Malus domestica and P. pyrifolia, is presented in Figure 2. Transpecific evolution is evident by the interspersion of alleles from different species throughout the tree. Use of parsimony to reconstruct the phylogeny gave essentially the same results. Under both methods of phylogenetic reconstruction, many of the nodes near the base of the tree are uncertain, but there is high support for many of the more recent nodes which subtend alleles from different taxa.

Neighbour-joining tree of S-alleles from Sorbus aucuparia (Sa), Crataegus monogyna (Cm), Malus domestica (Md), and Pyrus pyrifolia (Pp), rooted with allele S3 from Prunus avium (Rosaceae: Amygdaloideae). The tree was created using the K2P model implemented in PAUP* version 4.0b8. Support for nodes found in ≥70% of 1000 bootstrap replicates under neighbour-joining are indicated by the first number on internal branches. Numbers following commas indicate bootstrap support under parsimony (if >70%) using ACCTRAN optimization and ‘full heuristic’ searching. Sequence data for species other than S. aucuparia and C. monogyna are from Ishimizu et al (1998) and references therein.
Pairwise nucleotide diversity (mean ± s.e.) was 0.251 ± 0.010 in S. aucuparia and 0.280 ± 0.012 in C. monogyna, values lower than those observed in the Solanaceae (Table 2). Average terminal branch lengths in S. aucuparia and C. monogyna were significantly shorter than those in Solanum carolinense and Lycium andersonii, but were not significantly different from Physalis crassifolia (Table 2) whose S-alleles re-diversified following an historical bottleneck (Richman et al, 1996b). The depth of the genealogies, however, was much shallower in Rosaceae than in Solanaceae species (Table 2). This is because, even in P. crassifolia, multiple lineages predate the bottleneck event.
Patterns of average numbers of synonymous and nonsynonymous substitutions (uncorrected for the number of sites of each type) suggested purifying selection operating on the 5′ end of the amplified sequence up to the region C2 while diversifying selection might operate on much of the remaining sequence (Figure 3). We therefore analysed average Ds and Dn values separately for the first 26 amino acid positions and for position 27 to the 3′ end of the amplified sequence.

Average number of synonymous and nonsynonymous substitutions at each codon in pairwise comparisons of S-RNase sequences of Sorbus aucuparia and Crataegus monogyna. Note that values are not corrected for the number of sites. Underlined amino acids correspond to conserved region 2 (C2) of Ioerger et al (1990). The C1 and C5 regions are 5′ and 3′ of the amplified sequence, respectively. Gaps indicate the location of indels relative to other S-alleles in the sample.
For the first 26 codons, purifying selection was detected with greater per site values of synonymous (Ds) than nonsynonymous (Dn) diversity in both species (mean ± s.d.): S. aucuparia, Ds = 0.144 ± 0.043, Dn = 0.0456 ± 0.015, P < 0.05; C. monogyna, Ds = 0.209 ± 0.046, Dn = 0.066 ± 0.022, P < 0.05). These 26 codons include the highly conserved C2 region involved in RNA catalysis. However, even when the C2 region is excluded and the analysis is limited to the first 16 codons, Ds exceeds Dn by factors of 1.8 and 2.5 in S. aucuparia and C. monogyna, respectively. For positions 27 to the 3′ end of the amplified sequence, however, nonsynonymous diversity exceeded synonymous values, although the excess was statistically significant in only one of the species (S. aucuparia Ds = 0.204 ± 0.023, Dn = 0.265 ± 0.025, P < 0.05 (one-tailed); C. monogyna Ds = 0.274 ± 0.027, Dn = 0.278 ± 0.029, NS). A curious pattern is revealed by plots of Dsvs Dn for both species and for interspecific comparisons (Figure 4). At intermediate levels of sequence divergence (0.1 < Ds < 0.3) nonsynonymous site substitutions exceed synonymous ones. However, for either very close or distant comparisons, no excess of nonsynonymous substitutions is seen.

Pairwise synonymous (Ds) and nonsynonymous (Dn) substitutions per site for S-RNases from Sorbus aucuparia, Crataegus monogyna, and interspecific comparisons.
Discussion
Allele numbers and frequencies
The number of alleles estimated in S. aucuparia (24.4) and C. monogyna (26.7) fall within the range observed in previously studied species with single locus GSI. With the exception of populations of two Trifolium species, which often contain upwards of 100 alleles, populations of all other taxa with single locus GSI have been estimated to contain between 12 and 45 alleles (Lawrence, 2000). The estimated allele numbers are similar for the two species in this study, which might be expected given their similar life histories. Both species are bird-dispersed, insect-pollinated woody perennials and both tend to be opportunistic, colonising species of habitats in secondary successional stages. Richman et al (1996b) suggested that variation in allele numbers among species of Solanaceae might reflect variation in ecology with weedy species characterised by high population turnover having lower effective population sizes and lower allele numbers than species occupying undisturbed habitats. The allele number estimates reported here are intermediate between those reported for weedy (Physalis cinarescens, Witheringia maculata, and Solanum carolinense) and non-weedy (P. crassifolia, Lycium andersonii) Solanaceae (Richman, 2000; Richman and Kohn, 2000). The view that ecology is the major determinant of species allele number is controversial (Uyenoyama, 1997; Lu, 2001). More data are needed before the relative contributions of ecology and other factors can be evaluated.
Unequal frequencies of alleles have only rarely been demonstrated (Lawrence, 2000). This is surprising given that isoplethy is a property of populations at equilibrium, and equilibrium may not be the rule in nature. The apparent compliance to an equilibrium state might merely result from the low power of the traditional Mantel test (Lawrence, 2000) as well as the inherent low power of studies which survey such large numbers of alleles. Here we used Monte Carlo resampling to estimate the significance level of the Mantel statistic, which appears to be conservative when evaluated as a chi-square (Richman et al, 1996a). By this approach, allele frequencies were significantly unequal in C. monogyna, although the deviation had little effect on the estimate of allele number.
Unequal allele frequencies were not due to the multiple sampling of clones, since all individuals had unique genotypes. Unequal allele frequencies may result from many processes. In Papaver rhoeas, the S-locus is linked to a gene or genes affecting seed dormancy, which imposes an additional selection force and could be responsible for deviations from isoplethy (Lawrence and Franklin-Tong, 1994; Lane and Lawrence, 1995). Brooks et al (1996) also showed that limited pollen and seed dispersal, as well as variation in plant size could increase the variance of gene frequencies relative to expectations from simple frequency dependence. Apart from these ‘equilibrium’ processes, stochastic effects related to the founding of populations might equally be invoked to account for the deviation from isoplethy we observed in C. monogyna. The population we sampled is part of a hedge network among pastures, which is probably less than 100 years old. Since C. monogyna is a long-lived woody perennial, any effect of the founding of the population would likely remain detectable after such a short period of time.
We have confirmed previous observations from cultivated species of Rosaceae (Ishimizu et al, 1998) that intraspecific pairwise sequence divergence at the S-locus is low relative to the Solanaceae (Table 2). Average terminal branch lengths for the two Rosaceae species are not different from those found in P. crassifolia whose S-alleles underwent a burst of recent diversification (Richman et al, 1996b), but do differ from other Solanaceae (Table 2). While average terminal branch length is not independent of sampling effort, both the number of alleles sampled and the thoroughness of sampling are comparable for the Rosaceae and Solanaceae studied.
It is the depth of the genealogies of S-alleles from S. aucuparia and C. monogyna that differ markedly from those of Solanaceae. The Rosaceae species studied contain only alleles which appear to be of relatively recent origin while all Solanaceae, even those of P. crassifolia, contain polymorphism at the S-locus with much greater apparent time depth. Total sequence diversity in the Rosaceae studied is much lower than in Solanaceae because of reduced genealogy depth, even though levels of divergence between the most closely related alleles in the two families can be quite similar. Potential causes for this pattern will be analysed in greater detail in another study. One possibility is that recombination may be more frequent at the S-locus of the Rosaceae than the Solanaceae. Recombination reduces genealogy depth (Schierup and Hein, 2000; Uyenoyama, 2000), but whether reduction of apparent coalescence time by recombination is consistent with the maintenance of abundant transpecific polymorphism remains to be explored.
Patterns of selection at the S-locus of Rosaceae species are complex. First, different parts of the gene appear to be under different forms of selection. The first 26 codons sequenced in this study are subject to significant purifying selection as levels of nonsynonymous substitution are lower than levels of synonymous change. After codon 26, however, nonsynonymous change exceeds synonymous, though the excess is not statistically significant for C. monogyna. In particular, the region between amino acids 26 and 55 (Figure 3) which surrounds the position of the single intron appears to be most variable. This region has been termed ‘hypervariable’ in both the Solanaceae and Rosaceae and is believed to be a primary region for determining specificity (Ioerger et al, 1990; Kheyr-Pour et al, 1990; Matton et al, 1997; Ishimizu et al, 1998). However, Figure 3 also shows that the molecule remains quite variable at nonsynonymous sites for most of the remaining amplified region with relatively few highly conserved motifs. This suggests that sites involved in specificity differentiation may be scattered throughout the molecule, even outside regions considered hypervariable. The few conserved motifs in S-RNases represent either regions responsible for RNA catalysis or fixed structural features such as cysteine-cysteine bridges.
Excess of nonsynonymous substitutions is only apparent in comparisons involving intermediate levels of divergence (Figure 4). In comparison with most closely related and most divergent alleles, no excess was observed. Richman et al (1996b) ascribed the lack of excess nonsynonymous substitutions in comparison of widely divergent S-alleles of Solanaceae to the accumulation of silent substitutions over long periods of evolutionary time. If there are limits on the degree to which protein sequences can diverge and still function, synonymous substitutions will accumulate more rapidly than nonsynonymous ones as these limits are reached. The same process may apply here, even though the Rosaceae alleles are less divergent overall. The lack of excess nonsynonymous substitutions among most closely-related alleles is more difficult to explain since it is in these comparisons that diversifying selection might be expected to be most observable at the molecular level (and see Richman et al, 1995, 1996a, b).
In the current data, there are relatively few comparisons of closely-related alleles, and therefore any pattern (or lack of pattern) should be interpreted with caution. Nevertheless, unlike pairwise comparisons at intermediate levels of sequence divergence, each pairwise comparison of alleles that differ little in sequence is statistically independent, involving a separate sister pair of most-closely related alleles until Ds > 0.08. One possible interpretation of the observed pattern is that very closely related sequences do not represent different specificities and therefore show no excess of nonsynonymous substitutions. At present we have no molecular data from crosses involving the most similar pairs of putative S-alleles. However, little neutral variation within populations is expected to be found at the S-locus because the effective population size of each allele is small, equal to the effective size of the population divided by the number of S-alleles segregating within it (Clark, 1993). In addition, when the same allele was sequenced from more than one individual, no sequences that differed only in silent sites were discovered, suggesting little, if any, neutral variation is maintained. Instead, in the region analysed, all sequences differed by at least seven amino acids in Sorbus and eight amino acids in Crataegus. It is known from the Solanaceae that as few as four amino acid changes are sufficient to alter specificity (Matton et al, 1997). If closely related sequences indeed represent separate specificities, then the data may indicate that either the nature, or the detectability, of selection differs between the early, middle and late stages of divergence. More data collection among species of Rosaceae is warranted to see if the pattern observed here is general.
Comparative analysis of S-RNase diversification in different plant families holds much promise for discerning which features of evolution in these systems are repeatable, and which features vary with differences in species ecology, history, or differences in the molecular details of these systems in different taxonomic groups. While population S-allele numbers in Solanaceae and Rosaceae appear similar, the striking differences seen in levels of within-species sequence diversity warrant further work to elucidate how the same selective force, operating on apparently the same molecular system, gave rise to such different outcomes.
References
Brooks, RJ, Tobias, AM, Lawrence, MJ (1996). The population genetics of the self-incompatibility polymorphism in Papaver rhoeas. XI. The effects of limited pollen and seed dispersal, overlapping generations and variation in plant size on the variance of S-allele frequencies in populations at equilibrium. Heredity, 76: 367–376.
Campbell, JM, Lawrence, MJ (1981a). The population genetics of the self-incompatibility polymorphism in Papaver rhoeas. II. The number and frequency of S-alleles in a natural population (R106). Heredity, 46: 81–90.
Campbell, JM, Lawrence, MJ (1981b). The population genetics of the self-incompatibility polymorphism in Papaver rhoeas. I. The number and distribution of S-alleles in families from three localities. Heredity, 46: 69–79.
Clark, AG (1993). Evolutionary inferences from molecular characterization of self-incompatibility alleles. In: Takahata N, Clark AG (eds) Mechanisms of Molecular Evolution, Sinauer: Sunderland MA, USA pp 79–108.
Clark, AG, Kao, T-H (1991). Excess nonsynonymous substitution at shared polymorphic sites among self incompatibility alleles of Solanaceae. Proc Natl Acad Sci USA, 88: 9823–9827.
Edwards, SV, Hedrick, PW (1998). Evolution and ecology of MHC molecules: from genomics to sexual selection. Trends Ecol Evol, 13: 305–311.
Felsenstein, J (1995). PHYLIP (Phylogeny Inference Package). Release 3.572. Department of Genetics, University of Washington, Seattle, WA, USA.
Grime, JP, Hodgson, JG, Hunt, R (1988). Comparative Plant Ecology. A Functional Approach to Common British Species. Unwin Hyman: London, UK.
Ishimizu, T, Shinkawa, T, Sakiyama, F, Norioka, S (1998). Primary structural features of rosaceous S-RNases associated with gametophytic self-incompatibility. Plant Mol Biol, 37: 931–941.
Ioerger, TR, Clark, AG, Kao, T-H (1990). Polymorphism at the self incompatibility locus in the Solanaceae predates speciation. Proc Natl Acad Sci USA, 87: 9732–9735.
Kheyr-Pour, A, Bintrim, SB, Ioerger, TR, Remy, R, Hammond, SA, Kao, T-H (1990). Sequence diversity of pistil S-proteins associated with gametophytic self incompatibility in Nicotiana alata. Sex Plant Repro, 3: 88–97.
Korber, BT (2000). SNAP.p1 – Synonymous Nonsynonymous Analysis Program. Available at http://hiv-web.lanl.gov/SNAP/WEBSNAP/SNAP.html.
Kumar, S, Tamura, K, Jakobsen, IB, Nei, M (2001). MEGA2: Molecular Evolutionary Genetics Analysis software. Arizona State University, Tempe, Arizona, USA.
Lane, MD, Lawrence, MJ (1995). The population genetics of the self-incompatibility polymorphism in Papaver rhoeas. X. An association between incompatibility genotype and seed dormancy. Heredity, 75: 92–97.
Lawrence, MJ (2000). Population genetics of the homomorphic self-incompatibility polymorphisms in flowering plants. Ann Bot, 85: 221–226.
Lawrence, MJ, Franklin-Tong, VE (1994). The population genetics of the self-incompatibility polymorphism in Papaver rhoeas. IX. Evidence of an extra effect of selection acting on the S-locus. Heredity, 72: 353–364.
Lu, YQ (2001). Roles of lineage sorting and phylogenetic relationship in the genetic diversity at the self-incompatibility locus of Solanaceae. Heredity, 86: 195–205.
Mantel, N (1974). Approaches to a health research occupancy problem. Biometrics, 30: 355–362.
Matton, DP, Maes, O, Laublin, G, Xike, Q, Bertrand, C, Morse, D, et al (1997). Hypervariable domains of self-incompatibility RNases mediate allele-specific pollen recognition. Plant Cell, 9: 1757–1766.
McClure, BA, Haring, V, Ebert, PR, Anderson, MA, Simpson, RJ, Sakiyama, F, et al (1989). Style self-incompatibility gene products of Nicotiana alata are ribonucleases. Nature, 342: 955–957.
Nei, M, Gojobori, T (1986). Simple methods for estimating the number of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol, 3: 418–426.
O’donnell, S, Lawrence, MJ (1984). The population genetics of the self-incompatibility polymorphism in Papaver rhoeas. IV. The estimation of the number of alleles in a population. Heredity, 53: 495–507.
Paxman, GJ (1963). The maximum likelihood estimation of the number of self-sterility alleles in a population. Genetics, 48: 1029–1032.
Raspé, O (2000). Inheritance of the chloroplast genome in Sorbus aucuparia L. (Rosaceae). J Hered, 92: 507–509.
Raspé, O, Findlay, C, Jacquemart, A-L (2000a). Biological Flora of the British Isles: Sorbus aucuparia L. J Ecol, 88: 910–930.
Raspé, O, Saumitou-Laprade, P, Cuguen, J, Jacquemart, A-L (2000b). Chloroplast DNA haplotype variation and population differentiation in Sorbus aucuparia L. (Rosaceae: Maloideae). Mol Ecol, 9: 1113–1122.
Richman, AD (2000). S-allele diversity in Lycium andersonii: implications for the evolution of S-allele age in the Solanaceae. Ann Bot, 85: 241–245.
Richman, AD, Kao, T-H, Schaeffer, SW, Uyenoyama, MK (1995). S-allele sequence diversity in natural populations of Solanum carolinense (Horsenettle). Heredity, 75: 405–415.
Richman, AD, Kohn, JR (1999). Self-incompatibility alleles from Physalis: implications for historical inference from balanced genetic polymorphisms. Proc Natl Acad Sci USA, 96: 168–172.
Richman, AD, Kohn, JR (2000). Evolutionary genetics of self-incompatibility in the Solanaceae. Plant Mol Biol, 42: 169–179.
Richman, AD, Uyenoyama, MK, Kohn, JR (1996a). S-allele diversity in a natural population of Physalis crassifolia (Solanaceae) (ground cherry) assessed by RT-PCR. Heredity, 76: 497–505.
Richman, AD, Uyenoyama, MK, Kohn, JR (1996b). Allelic diversity and gene genealogy at the self-incompatibility locus in Solanaceae. Science, 273: 1212–1216.
Sassa, H, Nishio, T, Kowyama, Y, Hirano, H, Koba, T, Ikehashi, H (1996). Self-incompatibility (S) alleles of the Rosaceae encode members of a distinct class of the T2/S ribonuclease superfamily. Mol Gen Genet, 250: 547–557.
Schierup, MH, Hein, J (2000). Consequences of recombination on traditional phylogenetic analysis. Genetics, 156: 879–891.
Schneider, S, Roessli, D, Excoffier, L (2000). Arlequin: A software for population genetics data analysis. Release 2.000. Genetics and Biometry Lab, Dept. of Anthropology, University of Geneva, Switzerland.
Takahata, N (1990). A simple genealogical structure of strongly balanced allelic lines and transspecies evolution of polymorphism. Proc Natl Acad Sci USA, 87: 2419–2423.
Uyenoyama, MK (1997). Genealogical structure among alleles regulating self-incompatibility in natural populations of flowering plants. Genetics, 139: 1389–1400.
Uyenoyama, MK (2000). The evolution of breeding systems. In: Singh RS, Krimbas CB (eds) Evolutionary Genetics: From Molecules to Morphology. Cambridge University Press, Cambridge pp 300–316.
Vekemans, X, Slatkin, M (1994). Gene and allelic genealogies at a gametophytic self-incompatibility locus. Genetics, 137: 1157–1165.
Wang, X, Hughes, A, Tsukamoto, T, Ando, T, Kao, T-H (2001). Evidence that intragenic recombination contributes to allelic diversity of the S-RNase gene at the self-incompatibility (S) locus in Petunia inflata. Plant Physiol, 125: 1012–1022.
Wright, S (1939). The distribution of self-sterility alleles in populations. Genetics, 24: 538–552.
Wu, J, Saupe, SJ, Glass, NL (1998). Evidence for balancing selection operating at the het-c heterokaryon incompatibility locus in a group of filamentous fungi. Proc Natl Acad Sci USA, 95: 12398–12403.
Xue, Y, Carpenter, R, Dickinson, HG, Coen, ES (1996). Origin of allelic diversity in Antirrhinum S locus RNases. Plant Cell, 8: 805–814.
Yokoyama, S, Hetherington, LE (1982). The expected number of self incompatibility alleles in finite plant populations. Heredity, 48: 299–303.
Acknowledgements
This study was supported by funds to OR from the Belgian American Educational Foundation (BAEF 75 fellowship) and the Belgian National Fund for Scientific Research (FNRS), and National Science Foundation grants (DEB 9527834 and 0108173) to JRK. Sequencing was done by the UCSD Cancer Center DNA Sequencing Service. We thank AD Richman for providing his computer programmes to estimate population allele numbers and test for isoplethy.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Raspé, O., Kohn, J. S-allele diversity in Sorbus aucuparia and Crataegus monogyna (Rosaceae: Maloideae). Heredity 88, 458–465 (2002). https://doi.org/10.1038/sj.hdy.6800079
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.hdy.6800079
Keywords
This article is cited by
-
Characterization of 25 full-length S-RNase alleles, including flanking regions, from a pool of resequenced apple cultivars
Plant Molecular Biology (2018)
-
Identification, genealogical structure and population genetics of S-alleles in Malus sieversii, the wild ancestor of domesticated apple
Heredity (2017)
-
Genomic characterization of self-incompatibility ribonucleases in the Central Asian pear germplasm and introgression of new alleles from other species of the genus Pyrus
Tree Genetics & Genomes (2014)
-
Genetic variation and diversity among loquat accessions
Tree Genetics & Genomes (2014)
-
Evaluation of candidate F-box genes for the pollen S of gametophytic self-incompatibility in the Pyrinae (Rosaceae) on the basis of their phylogenomic context
Tree Genetics & Genomes (2011)
