
Abstract
The self-incompatibility system of flowering plants is a classic example of extreme allelic polymorphism maintained by frequency-dependent selection. We used primers designed from three published Antirrhinum hispanicum S-allele sequences in PCR reactions with genomic DNA of plants sampled from natural populations of Antirrhinum and Misopates species. Not surprisingly, given the polymorphism of S-alleles, only a minority of individuals yielded PCR products of the expected size. These yielded 35 genomic sequences, of nine different sequence types of which eight are highly similar to the A. hispanicum S-allele sequences, and one to a very similar unpublished Antirrhinum S-like RNase sequence. The sequence types are well separated from the S-RNase sequences from Solanaceae and Rosaceae, and also from most known ‘S-like’ RNase sequences (which encode proteins not involved in self-incompatibility). An association with incompatibility types has so far been established for only one of the putative S-alleles, but we describe evidence that the other sequences are also S-alleles. Variability in these sequences follows the pattern of conserved and hypervariable regions seen in other S-RNases, but no regions have higher replacement than silent diversity, unlike the results in some other species.
Similar content being viewed by others


Introduction
Self-incompatibility (SI) is a genetic mechanism in flowering plants that prevents self-fertilisation, by enabling the pistil to reject pollen from genetically related individuals, and is controlled by multi-allelic the S-loci (de Nettancourt, 1977). In gametophytic self-incompatibility systems that have a single S-locus, identity of the allele occurring in a pollen grain with either of the alleles in a recipient pistil is sufficient to prevent fertilisation. Molecular studies of the pistil incompatibility locus in three plant families, Solanaceae, Rosaceae and Scrophulariaceae, have shown that the pistil S-proteins are members of a class of extracellular basic glycoproteins with ribonuclease activity, the S-RNases (reviewed by Kao and McCubbin, 1996, 1997; Sassa et al, 1996; Xue et al, 1996).
Other genes with similar structure to S-RNases, but not linked to the S-locus (the ‘S-like RNases’), exist in various plant families including the Solanaceae (Jost et al, 1991; Löffler et al, 1993; Kuroda et al, 1994; Dodds et al, 1996; Lers et al, 1998) and Rosaceae (Norioka et al, 1996; Ma and Oliveira, 2000). Recently, two putative S-like RNase sequences have been obtained from Antirrhinum mollissimum (YB Xue, unpublished; GenBank accession numbers AJ315591 and AJ315592). The S-like RNases, are believed not to be involved in self-incompatibility, but to function in other processes involving RNA turnover (Löffler et al, 1993; Bariola et al, 1994; Lers et al, 1998; Ye and Droste, 1996; Gausing, 2000).
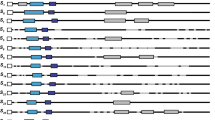
Figure 1 shows a schematic diagram of the known features of S-RNases and similar sequences. Information on S-alleles in the Scrophulariaceae is currently based on only three cDNA sequences from Antirrhinum hispanicum (Xue et al, 1996). Genetic evidence showed linkage to the S-locus and the sequences were shown to be specifically expressed in styles, with transcript levels that peak at anthesis (Xue et al, 1996). These three sequences contain four short highly conserved regions and two hypervariable regions, HVa and HVb (Xue et al, 1996). Four of the conserved regions (C1, C2, C3 and C5 in Figure 1) are in similar positions to those previously noted in the S-RNases of Solanaceae (Kao and McCubbin, 1996) and Rosaceae (Norioka et al, 1996; Sassa et al, 1996; Ushijima et al, 1998). C2 and C3 are also recognised in the RNases of the fungus Rhizopus niveus (Green, 1994), and two of the three histidine residues required for catalytic activity are in these regions (Ohgi et al, 1992; see also Figure 3). The two HV regions are also found in S-RNases of Solanaceae, but only HVa is recognised in the S-RNases so far studied in the Rosaceae (Norioka et al, 1996; Sassa et al, 1996; Ushijima et al, 1998). The HV regions are the most hydrophilic regions of the S-proteins (Kao and McCubbin, 1996; Broothaerts et al, 1995; Xue et al, 1996), and may be on the external surface of the protein. Given their high degree of variability, they are candidates for the regions determining S-allele specificity. Alternatively, they could merely be tolerant to sequence variation (Zurek et al, 1997).

Features of S-RNase and S-like genes of different kinds from Solanaceae, Rosaceae and Scrophulariaceae (S-RNases only), showing the regions of conserved sequence (grey bars), the hypervariable regions in the S-allele sequences (striped bars), and the known introns (black bars).

Numbers of different amino acids at each position in the Antirrhinum and Misopates S-RNases. Only one sequence of each type is shown. The C1-C3 conserved regions and the hypervariable (HV) found in S-RNases of species from other plant families, and the intron positions, are indicated.
Two different types of molecular experiments have been used to test the importance of the hypervariable regions in determining specificity. Swapping entire regions between very different S-alleles led to the conclusion that in Petunia inflata and Nicotiana alata (both members of the Solanaceae) the S-RNase recognition function is not localised to a specific domain in any of the alleles studied (Kao and McCubbin, 1997; Zurek et al, 1997), but the HV regions control specificity differences between some S-alleles. Replacing just the hypervariable region of one S. chacoense S-allele with that a different allele (with four amino acid differences in this region and six elsewhere in the protein) changed the pistil incompatibility reaction to that of the allele from which the introduced sequence was taken (Matton et al, 1997). Exchanging a single amino acid in the HVa region of one S. chacoense S-allele with that of a different allele produces a new, dual-specificity S-allele, which simultaneously rejects pollen tubes with two different incompatibility types (Matton et al, 1999).
Studying the frequencies of amino acid and silent changes in different regions of the gene may also help to identify regions that are important for specificity. In the highly polymorphic class I major-histocompatibility-complex (MHC) loci, non-synonymous nucleotide substitutions occur significantly more frequently than synonymous ones at sites in the binding cleft of the antigen recognition regions (Hugheset al, 1990). Measuring non-synonymous and synonymous substitutions between alleles as their per site values, Ka and Ks, the finding of Ka/Ks ratios greater than unity suggests the operation of balancing selection (Hughes et al, 1990). This does not, however, identify which individual amino acids are under balancing selection. S-alleles are maintained in natural populations for long time periods by balancing selection due to the frequency-dependent advantage of rare alleles (Wright, 1939). Regions very closely linked to the target(s) of selection may thus accumulate sequence differences, and Ka/Ks ratios approaching unity may arise if selective constraints are low in these regions of the protein. In Solanum, however, Ka/Ks ratios exceed 1 in the HV regions of pairs of alleles that are similar in sequence, though the ratio declines at high Ks (Richman et al, 1996, Richman and Kohn, 1999). In the Rosaceae, Ka/Ks exceeds one in four putatively selected (‘PS’) regions of P. pyrifolia and Malus × domestica S-alleles, one of them spanning the HVa region (Ishimizu et al, 1998). The PS1 region includes the HV region of Rosaceae, and PS1 and PS2 correspond to HVa and HVb in the S-RNases of Solanaceae.
In order to do similar analyses in the Scrophulariaceae, and test whether there is evidence that their HV regions behave as expected for the targets of selection, further alleles from natural populations must be studied. We here describe genomic sequences (including both HVa and HVb regions, C2 and C3, and an intron) of putative S-alleles from populations of the genera Antirrhinum and Misopates.
Materials and methods
Plant material and DNA extraction
The plants studied were collected in the field in Portugal (Table 1). They were from single populations of three self-incompatible Antirrhinum species (A. graniticum, A. molle and A. meonanthum), two subspecies of A. majus (the partially self-incompatible A. majus subsp. linkianum, two largely self-compatible populations of subspecies cirrhigerum, denoted Ave and Gala in the table, and one largely self-incompatible population of this subspspecies, denoted Muel), one self-compatible Misopates orontium population and one self-compatible Misopates calycinum population. Genomic DNA was extracted from leaves of individual plants collected in the field, using the method of Ingram et al (1997).
Crosses in A. graniticum
Two plants (graniticumB1–1 and graniticumB1–2) were grown in the glasshouse from seeds of one fruit of a wild A. graniticum individual (graniticumB1). Another wild plant (graniticumB4) in the same population was the maternal parent of three other plants (graniticumB4–1, graniticumB4–2, and graniticumB4–3). Four crosses were performed between these five plants (all of which were highly cross-compatible in both reciprocal crosses):
graniticumB1–1 × B4–1
graniticumB1–1 × B4–2
graniticumB1–1 × B4–3
graniticumB1–2 × B4–3
A total of 21 plants from seeds of these crosses were used in reciprocal cross-pollinations to determine their incompatibility groups. Each pollination was repeated at least three times. Unfortunately, details of the ancestry of the 21 individuals were not recorded.
PCR amplification and sequencing
Primers were designed based on the conserved regions in the cDNA sequences of the three A. hispanicum S -alleles, S2, S4 and S5 (GenBank accession numbers X96465, X96466, and X96464 respectively). Two primers were designed for region C1: ants2s3F (position 108 and 112, relative to the start codon of the S2 and S5 cDNA sequences; 5′ AAATTGGTTCT(TC)CAATGG 3′) and ants4F (position 109 of the S4 sequence; 5′ AAGCTAGTTCTCCAATGG 3′). Primer ants2s3R (position 560 of the S2 and S5 sequence; 5′AACCTTCA(CG)(CG)GCATTTCA 3′) and ants4R (position 560 of the S4 sequence; 5′ ATAACTTTGAGCACATTTCA 3′) were designed for the conserved motif LKC, which is just 5′ to region C5. Primers for the S4 sequences are specific for S4 sequences only, and no matching sequences are present in the S2 and S5 sequences. The other primers match sequences present in both S2 and S5 sequences, and may therefore be expected to amplify at least these two types of alleles, and perhaps others as well. Because of the size of the S4 genomic region analysed (see below), additional internal primers are required to sequence both strands, and additional primers were therefore designed. Sequences were obtained using the following primer pairs: ants4F with the additional primer ants4–1 (based on the sequences obtained here; 5′ GATTGCTCAGAACTTTTTAC 3′), and additional primer ants4–2 (designed for position 272 of the S4 cDNA sequence; 5′ TCTCTGCTCTATGTCTGTAT 3′) with ants4R. Standard amplification conditions were 35 cycles of denaturation at 94°C for 30 s, primer annealing at 49°C for 30 s, and primer extension at 72°C for 2 min.
To test for homogeneity of the PCR products, which could reveal that more than a single loci had been amplified, we digested with four 4 bp-cutting restriction enzymes (AciI; AluI, RsaI, and DdeI). When this revealed numbers and/or sizes of bands that are incompatible with that of the sequence from which the primers were designed, the product was cloned using the TA cloning kit (Invitrogen) and multiple colonies were screened until several of each type were found. At least three different colonies were then sequenced, to obtain a consensus sequence.
Both forward and reverse sequences were obtained, using an Applied Biosystems model 377 DNA sequencing system with the ABI PRISM BigDye cycle-sequencing kit (Perkin Elmer), and specific primers or the primers for the M13 forward and reverse priming sites of the pCR2.1 vector. Almost all the sequences reported here were obtained by direct sequencing of PCR products, without cloning, as each primer set usually yielded only one sequence.
Analyses of sequences
The DNA sequences were deposited in GenBank (accession numbers AY013879–AY013914). The nucleotide sequences of the putative coding regions to be compared were aligned using ClustalX v. 1.64b (Thompson et al, 1997), and minor manual adjustments were performed using SeqPup v. 0.6f. The numbers of synonymous and nonsynonynous differences between pairs of sequences were calculated using the DnaSP software (Rozas and Rozas, 1997). Neighbour-joining trees were generated with MEGA version 1.01 (Kumar et al, 1994).
Results
Analysis of A. graniticum crosses
The 21A. graniticum plants tested fall into seven self-incompatibility groups (I–VII, Table 2). All groups were reciprocally cross-compatible with one another, except for 20 pollinations (out of 548, 3.6%; Table 2) between different groups that yielded no seeds, probably because of damage during handling of the flowers used in the pollinations. Within groups, all plants were cross- incompatible, with the exception of a total of three pollinations in groups II and V, in which more than 10 seeds were obtained; these are likely to be the result of contamination with pollen from other plants.
Genomic DNA of these 21 individuals was used in PCR amplifications using two different pairs of primers. The primer pair ants4F and ants4R did not amplify from any of these 21 plants. Primer pair ants2s3F and ants2s3R yielded a product about 360 bp long (smaller than the expected amplification product) in all 21 plants, probably a non-specific amplification product. In addition, a 658 bp amplification product was obtained with these primers from all individuals of groups II, III and IV, but not from any plants of any of the other groups. The presence of the 658 bp band is highly significantly associated with incompatibility groups, by a contingency test (χ2 = 21, with 7 df). Because the expected numbers in some cells are small, significance was tested by randomising the data, preserving the row and column totals. No χ2 value as high as that observed for the actual data was observed in 10 000 randomisations.
The patterns of the 658-bp PCR product were identical in the eight individuals that amplified, with four different restriction enzymes, and this product was sequenced for one individual. The highest amino acid similarity by BlastX search is with the published A. hispanicum S5 sequence (58% amino acid identity). This product may therefore be a new functional Antirrhinum S-allele, which we shall denote S6. There is an insertion of 186 bp, between amino acids 50 and 51 in the S6 sequence, relative to the Antirrhinum S5 cDNA sequence (Figure 2). This is probably an intron, and indeed the A. hispanicum S2-RNase genomic sequence (GenBank accession number AJ300474) has an intron in the same position, when compared with the cognate cDNA sequence.

Neighbour-joining tree showing the relationships among the coding sequences of the Antirrhinum and Misopates S-RNases and those of the three published Antirrhinum S-allele cDNAs. The names of the different sequence types are as in Table 1, and the lengths of the putative introns are shown in parentheses. The numbers by the nodes of the tree are bootstrap values greater than 68%.
At least one intron is expected, since the S-alleles of S-RNases of Solanaceae and Rosaceae usually have a single intron (in the HVa region, from 87 bp to 120 bp, and 138 bp to 1100 bp, respectively, in sequences from these two families; Saba-El-Leil et al, 1994; Matton et al, 1995; Broothaerts et al, 1995). Prunus (Rosaceae) S-RNases have at least two introns (in P. avium, one 5′ to the C1 region, and one in HVa; Tao et al, 1999), and a sequence from P. dulcis (GenBank accession number AF157008) is similar (Figure 1). To determine consensus splice site sequences in A. majus, we examined 31 introns from eight A. majus nuclear genes (chs, X03710; TAP1, X57295; DEFA, X62810; fil2, X76995; far, J239057; squamosa, X63701; fil1, X57296; and globosa, X68831). In agreement with these other Antirrhinum genes, and with the A. hispanicum S2-RNase, the sequences deduced to be the intron/exon boundaries in our sequences all have 5′ GT. . . and AG 3′ sequences.
Distribution of putative S-alleles in plants sampled from natural populations
The primer pair ants2s3F and ants2s3R was also used in PCR amplifications of genomic DNA of four Antirrhinum and two Misopates species. From a total of 170 plants sampled in the wild, the 360 bp product mentioned above again amplified from plants of all different populations and species, but products of a size likely to represent S-alleles amplified from only 30 individuals (Table 1).
When the PCR products were digested with restriction enzymes, the amplification product of only one individual (molle4) was heterogeneous (Table 1). This individual's PCR amplification product was cloned. Digestion of 20 clones with restriction enzymes revealed only two types of clones (A has only one AciI restriction site, while B has two AciI sites and one RsaI site). Several clones of each type were sequenced. Clone type B is the same as the S6 sequences, except for a 1 bp indel in the putative intron region, and clone type A is a new S-RNase sequence, which was also found in two A. meonanthum, two A. molle and two A. graniticum plants (Table 1 and Figure 2).
Three of the sequences from A. graniticum (graniticumB4, graniticumB6 and graniticumB29) were identical to the S6 allele detected in the plants of A. graniticum incompatibility groups II, III, and VII classified by hand-pollinations. One sequence (graniticumB15), which is the most divergent from all other sequences obtained (Figure 2), had 99% identity to an unpublished putative S-like sequence recently obtained from Antirrhinum mollissimum (YB Xue, personal communication; GenBank accession number AJ315592). BlastX search of the remaining 26 sequences gave the highest amino acid similarity (63% to 99% amino acid identity) with the published Antirrhinum S2 or S5 allele sequences.
The same 170 individuals were tested with primer pair ants4F and ants4R. These amplified a PCR product in only four individuals, from two different Antirrhinum species (Table 1). The four products are of similar size (1386 bp in graniticumB14 and 1389 bp in cirrhigerumGala9, cirrhigerumGala11, and cirrhigerumGala14). Digestion with restriction enzymes showed that all four amplification products are homogeneous. Sequences were obtained, and BlastX search showed 98% amino acid identity with the published S4 Antirrhinum sequence, as might be expected given that the primers used in these amplifications were designed based on the S4 sequence, and may be specific for this sequence.
In all these sequences, a putative intron was deduced by comparison with the S-allele cDNA sequences in GenBank. Its position is always between amino acids 50 and 51 (Figure 3), but its size varies from 132 to 956 bp (Figure 2). The intron/exon boundaries are conserved and again agree with the deduced consensus splice sites of Antirrhinum nuclear genes. The putative intron sequences all contain several stop codons if they are translated in reading frame with the rest of the sequence, and Blast searches of these sequences reveals no similarity with any sequence in GenBank.
Variability of the Antirrhinum and Misopates putative S-allele coding sequences
The 33 Antirrhinum and two Misopates coding sequences now determined fall into nine different sequence groups, of which five (S6, A, B, C, and S4) were found as almost identical sequences in multiple individuals, including plants from different species (Figure 2). Three unique sequences (the sequences in plants graniticumB27, meonanthum4, and graniticumB15) were also found, in addition to one S5, a sequence originally found in A. hispanicum; no sequence similar to S2 of A. hispanicum was present in our samples. Two individuals (molle4 and cirrhigerumGala9) had two different sequences and may be heterozygotes.
Figure 2 shows the relationships among the coding sequences reported here together with the three published Antirrhinum S-allele cDNAs (Xue et al, 1996). Between the different sequence groups, the introns differ greatly, and cannot be aligned between the different sequences, and Ks values are too high to estimate accurately. The published S4Antirrhinum sequence clusters with the graniticumB14 and three cirrhigerumGala sequences (group S4 in Figure 2). The coding sequences of the S4 group differ at three nucleotide sites. Two differences (at positions 145 and 342) are fixed between our four sequences and the published S4 Antirrhinum sequence, and the third difference is between graniticumB14 and the S4 Antirrhinum sequence (position 323). The Antirrhinum S5 sequence clusters with the linkianum 1 sequence, which differs from it by two positions in the coding region (positions 273 and 546).
Within each of the sequence groups, the coding regions have low variability, both within the species and between them, though our samples within species are small (Table 1 and Figure 2). Among the five sequences of the S6 group, from two different species, the only variant was a length difference of one nucleotide in the intron between the molleB sequence and the four from A. granticium. Five other sequence groups included at least two genomic sequences, so that the intron could be analysed. Diversity per intron site was low, and all intron variants, except for two among the three A. molle group A sequences, were between species. Only 92–95 silent coding region sites are available from all the groups sequenced, but there are about 340 non-synonymous sites. Silent site variability within groups was also low, even though the groups often include sequences from more than one species. Within three groups, there were no silent site variants (S6, mentioned above, plus the seven group A and five B sequences). All silent variants in other groups were differences between species, except for the two group C sequences from the self-compatible M. orontium population, which differ at many synonymous sites; one sequence is identical to the cirrhigerumGala9 sequence, also from a self-compatible population. For S5 (two sequences, from two different species), silent site divergence was 1.1% and there was also one amino acid replacement difference.
Amino acid replacement variants were also found within all other sequence groups except S6, also largely between species. No replacement variants were found among the three S4 group sequences from A. majus cirrhigerum, all from one self-compatible population of this subspecies, but the S4 sequences from other species had replacement differences (Figure 2). For three sequence groups (A, B and C), comparisons between and within species are available. Among group B, all replacement variants, like the non-synonymous ones, were between the two species in which this sequence type was found (Figure 2). There were also no non-synonymous variants among the five A. molle group C sequences, and in A. majus ssp.cirrhigerum the only difference was between two sequences from different populations. However, three non-synonymous variants were found between the two group C sequences from the M. orontium population, and one between the two A group sequences from the A. meonanthum population (though none was found among the six other A group sequences, from three species; Figure 2).
Comparison of the Antirrhinum and Misopates putative S-allele sequence with S-RNases of other plants
The sequences obtained here have features similar to those of other S-RNases, in the region analysed (Figure 3). The regions that are conserved among the sequences reported here correspond to the portion of the C1 region that is included in our sequences, plus regions C2 and C3 of S-RNases from Solanaceae and Rosaceae. Two regions that are conserved in S-RNases of other plant families (regions C4 of Solanaceae, corresponding to amino acids 109 to 116 in Figure 3, and RC4 of Rosaceae, corresponding to amino acids 114 to 126 in Figure 3) are not conserved in the three A. hispanicum cDNA sequences (Xue et al, 1996) nor in our sequences. A total of 32 amino acid residues are conserved among all sequences from species of Scrophulariaceae (17 outside the conserved regions; Figure 3). Five of these sites (5, 12, 39, 63, and 102) are also conserved in the S-RNase sequences from the Solanaceae and Rosaceae (Richman et al, 1997, Ushijima et al, 1998), four of them (amino acids 18, 103, 110, and 116) in sequences from the Solanaceae, and three (amino acids 64, 81 and 86) in sequences from the Rosaceae. Given the large amino acid divergence between the sequences, these are likely to be functionally important amino acids.
The Antirrhinum putative S-RNase sequence groups are very divergent from one another, with high Ks values (0.90 ± 0.36) even for non-HV regions (Figure 4). The HVa and HVb hypervariable regions observed in S-RNases from other plant families are, however, also the regions with most variability in the Antirrhinum and Misopates sequences (Figure 3). These regions include several insertion/deletion (indel) differences between the sequence groups, as well as many silent site differences (Figure 4). They also have more amino acid replacements than non-HV regions, but not significantly so (Ka values, and their standard deviations, for the two kinds of regions are estimated as 0.67 ± 0.25 for HV, vs 0.41 ± 0.16 for non-HV regions).

Ka/Ks values for pairwise comparisons of the putative S-RNase sequences from several Antirrhinum and Misopates species. Ka and Ks values are the numbers of nonsynonymous and synonymous substitutions per site between sequences of different groups, and the thick line shows Ka/Ks = 1. The filled symbols show the results using the entire sequence region analysed, and the open symbols are excluding the conserved regions C1, C2 and C3.
Discussion
Evidence that the sequences are S-alleles
With the sequences reported here and the three found in A. hispanicum (Xue et al, 1996), a total of 10 Scrophulariaceae potential S-RNase sequence types are now known. Before we can analyse these data and investigate questions of interest (see Introduction), we need to know whether the sequences represent S-alleles. All the sequences have the features and patterns of diversity typical of S-RNases (Figure 1), including a single intron in the HVa region, and some of them are extremely similar to the Antirrhinum S4 and S5 alleles (Xue et al, 1996), but this does not rule out the possibility that they are S-like RNases.
The S-like RNases from Solanaceae and Rosaceae have introns in similar positions to those in S-RNase sequences (Ma and Oliveira, 2000). Some have a single intron in the region corresponding to HVa of S-RNases, as do most S-RNases known from the species from which they were obtained. One cannot, therefore, distinguish between S-like RNases and S-alleles by their sequences alone. Other S-like RNases are like the S-RNases of P. avium in having an additional intron in the C1 region, and some have a third intron 3′ to C3. Intron positions are not known for the two A. mollissimum S-like RNases, as only cDNA sequences are so far available (YB Xue, unpublished; GenBank accession numbers AJ315591 and AJ315592).
Ideally, to show that sequences are good candidates for S-alleles, there should be evidence of linkage to the S-alleles segregating in families or populations. This is rarely available for natural populations, though data are starting to be obtained for species in the Solanaceae (Wang et al, 2001). We found complete association between the presence of one putative S-allele sequence and incompatibility groups, in a set of plants from four related sibships. Although these data are not equivalent to a definitive test of linkage to the S-locus, they support the view that our sequences are not merely S-like paralogues.
Furthermore, the PCR primers tested here amplified similar sequences from different species, but from only a minority of individuals of any population (Table 1). Such results are expected for the S-locus; bands are not obtained for every individual, even under low stringency conditions, because the sequences of different alleles are highly diverged (eg, Richman et al, 1996). In contrast, PCR amplification experiments and Southern blot hybridisations using sequences of specific S-like RNase loci generally detect a band in every individual, since the sequences of given loci are conserved (Dodds et al, 1996; Norioka et al, 1996; Ma and Oliveira, 2000).
Genealogical analyses of the RNases of Scrophulariaceae, Solanaceae and Rosaceae
The relationships between the S-allele sequences from Scrophulariaceae, Solanaceae and Rosaceae are shown in Figure 5. The figure is based on amino acid differences between the most divergent S-allele lineages within each plant family (Richman et al, 1997), plus the 14 known S-like RNase sequences from various species of Solanaceae and Rosaceae, some of which were not available for the previous analyses (Xue et al, 1996; Richman et al, 1997; Ushijima et al, 1998). As was found previously, most of the Antirrhinum sequences determined so far (including the A. mollissimum putative S-like RNase sequence, AJ315592) cluster together as a sister group to S-allele sequences. S-like RNase sequences of Solanaceae and Rosaceae, however, mostly form a distinct cluster, which includes the A. mollissimum S-like RNase, AJ315591 (Figure 5; the four exceptions, indicated by asterisks, are the A. mollissimum sequence, AJ315592, the Petunia inflata RNase X2 sequence and two sequences from N. alata). This suggests that most, but maybe not all, of the S-like RNase genes probably diverged before the most recent common ancestor of the taxa from which they were sampled. The fact that sequences of putative S-RNases from the three different plant families form separate clusters suggests that the variability within the three families does not pre-date the families themselves. Thus, either self-incompatibility evolved independently in each of them, or else variants have been replaced by new ones that have arisen within the families.

Neighbour-joining tree based on amino acid differences between S-allele lineages and S-like RNase sequences from various species of Solanaceae and Rosaceae, and the Antirrhinum and Misopates S-RNase sequences. The numbers by the nodes of the tree are bootstrap values greater than 68%.
Another source of evidence on whether our sequences are allelic variants at a single (S) locus, or could come from several loci (including S-like RNase genes) would be to test for linkage disequilibrium. Variants in highly polymorphic sequences pooled from non-allelic loci should be in linkage disequilibrium, whereas this is not expected for allelic variants with sufficient recombination (Awadalla and Charlesworth, 1999). The presence of introns in S-RNase genes makes it difficult to do this test, however, because there are alignment problems due to indel variants. Moreover, even if a reliable alignment could be obtained, the distances between polymorphic sites would not be the same in different sequences. We have therefore not used this approach.
S-allele sequence evolution
In contrast to the results from species in the Solanaceae (Richman et al, 1996; Richman and Kohn, 1999), Ka/Ks values are high, but do not exceed unity; the average Ka/Ks value over the sequence region analysed here (Figure 4) is 0.460 ± 0.083, or 0.534 ± 0.081 excluding the conserved regions C1, C2 and C3 (Figure 4). This remains true for just the region spanning amino acids 27 to 87 studied by Richman et al (1996). This analysis therefore provides no compelling evidence for diversifying selection. Nevertheless, if they are allelic, the great sequence differences between them indicates that they must have been polymorphic for a long time, suggesting balancing selection acting on the locus. This is supported by the trans-specific polymorphism of these putative S-alleles, though hybridisation between species cannot be excluded. The absence of very high Ka/Ks values in the S-RNase sequences from Scrophulariaceae studied here could be due to a greater age of the S-alleles. In the sequences from Solanaceae, high Ka/Ks values were found only between alleles that are not highly diverged (Richman et al, 1996), possibly because the existence of conserved sites places an upper limit on protein sequence divergence, but not silent site divergence (Tanaka and Nei, 1989).
More detailed analysis of these sequences is not yet worthwhile, as the sequences of many alleles are probably not yet known, given the failure to amplify sequences from many individuals, and the fact that those from which putative S-alleles can be amplified rarely yielded two sequences. It is, however, interesting that there is no sign from the region of sequence analysed that the nine putative S-allele sequences from self-compatible populations (seven from the cirrhigerumGala population of A. majus, and two, both of sequence group C, from M. orontium) are pseudogenes. The intron splice sites are also conserved, and there are no interruptions of the open reading frame. It is unknown why these populations are self-compatible, and whether they have functional pistil incompatibility reactions. It is generally accepted that the genera Antirrhinum and Misopates are closely related, and analyses of other nuclear gene sequences support this (Vieira et al, 1999). Thus, even if the S-alleles of these populations are non-functional due to mutations elsewhere in the gene, there has probably been little time for accumulation of further mutations.
There were, however, a number of amino acid replacement variants in the M. orontium sequences, and also between the Gala and Muel populations of A. majus ssp. cirrhigerum, unlike the situation for other sequence groups that could be compared within species (except for one such variant in A. meonanthum). The numbers of sequences available at present are not large, and more data would be valuable. These differences could be associated with the loss of incompatibility in these populations, but there is also no evidence at present, and self-compatibility could well be due to mutations of other genes involved in incompatibility (Brieger, 1935; Tseng, 1938). It will be interesting in the future to test this, and also whether the amino acid differences between the A group sequences within A. meonanthum are associated with the same or different SI types.
References
Awadalla, P, Charlesworth, D (1999). Recombination and selection at Brassica self-incompatibility loci. Genetics, 152: 413–425.
Bariola, PA, Howard, CJ, Taylor, CB, Verburg, MT, Jaglan, VD, Green, PJ (1994). The Arabidopsis ribonuclease gene RNS1 is tightly controlled in response to phosphate limitation. Plant J, 6: 673–685.
Brieger, FG (1935). The inheritance of self-sterility and the peloric flower shape in Antirrhinum. Genetica, 17: 385–408.
Broothaerts, W, Janssens, GA, Proost, P, Broekaert, WF (1995). cDNA cloning and molecular analysis of two self-incompatibility alleles from apple. Plant Mol Biol, 27: 499–511.
de Nettancourt, D (1977). Incompatibility in Angiosperms. Springer-Verlag, Berlin.
Dodds, PN, Clarke, AE, Newbigin, E (1996). Molecular characterisation of an S-like RNase of Nicotiana alata that is induced by phosphate starvation. Plant Mol Biol, 31: 227–238.
Gausing, K (2000). A barley gene (rsh1) encoding a ribonuclease S-like homologue specifically expressed in young light-grown leaves. Planta, 210: 574–579.
Green, PJ (1994). The ribonucleases of higher plants. Annu Rev Plant Physiol Plant Mol Biol, 45: 421–445.
Hughes, A, Ota, T, Nei, M (1990). Positive Darwinian selection promotes charge profile diversity in the antigen-binding cleft of class I major-histocompatibility complex molecules. Mol Biol Evol, 76: 515–524.
Ingram, GC, Doyle, S, Carpenter, R, Schultz, EA, Simon, R, Coen, ES (1997). Dual role for fimbriata in regulating floral homeotic genes and cell division in Antirrhinum. EMBO J, 16: 6521–6534.
Ishimizu, T, Endo, T, Yamaguchi-Kabata, Y, Nakamura, KT, Sakiyama, F, Norioka, S (1998). Identification of regions in which positive selection may operate in S-RNase of Rosaceae: implication for S-allele-specific recognition sites in S-RNase. FEBS Lett, 440: 337–342.
Jost, W, Bak, H, Glund, K, Terpstra, P, Beintema, JJ (1991). Amino acid sequence of an extracellular, phosphate-starvation-induced ribonuclease from cultured tomato (Lycopersicon esculentum) cells. Eur J Biochem, 198: 1–6.
Kao, T-H, McCubbin, AG (1996). How flowering plants discriminate between self and non-self pollen to prevent inbreeding. Proc Natl Acad Sci USA, 93: 12059–12065.
Kao, T-H, McCubbin, AG (1997). Molecular and biochemical bases of gametophytic self-incompatibility in Solanaceae. Plant Physiol Biochem, 35: 171–176.
Kumar, S, Tamura, K, Nei, M (1994). MEGA- molecular evolutionary genetics analysis software for microcomputers. Comp Appl Biosci (CABIOS), 10: 189–191.
Kuroda, S, Norioka, S, Mitta, M, Kato, I, Sakiyama, F (1994). Primary structure of a novel stylar RNase unassociated with self-incompatibility in tobacco plant, Nicotiana alata. J Protein Chem, 13: 438–439.
Lers, A, Khalchitski, A, Lomaniec, E, Burd, S, Green, PJ (1998). Senescence-induced RNases in tomato. Plant Mol Biol, 36: 439–449.
Löffler, A, Glund, K, Irie, M (1993). Amino acid sequence of an intracellular, phosphate-starvation-induced ribonuclease from cultured tomato (Lycopersicon esculentum) cells. Eur J Biochem, 214: 627–633.
Ma, RC, Oliveira, MM (2000). The RNase PD2 gene of almond (Prunus dulcis) represents an evolutionarily distinct class of S-like RNase genes. Mol Gen Genet, 263: 925–933.
Matton, DP, Mau, SL, Okamoto, S, Clarke, AE, Newbigin, E (1995). The S-locus of Nicotiana alata: genomic organization and sequence analysis of two S-RNase alleles. Plant Mol Biol, 28: 847–858.
Matton, DP, Maes, O, Laublin, G, Xike, Q, Bertrand, C, Morse, D et al (1997). Hypervariable domains of self-incompatibility RNases mediate allele-specific pollen recognition. Plant Cell, 9: 1757–1766.
Matton, DP, Luu, DT, Xike, Q, Laublin, G, O’brien, M, Maes, O et al (1999). Production of an S RNase with dual specificity suggests a novel hypothesis for the generation of new S alleles. Plant Cell, 11: 2087–2097.
Norioka, N, Norioka, S, Ohnishi, Y, Ishimizu, T, Oneyama, C, Nakanishi, T et al (1996). Molecular cloning and nucleotide sequences of cDNAs encoding S-allele specific stylar RNases in a self-incompatible cultivar and its self-compatible mutant of Japanese pear, Pyrus pyrifolia Nakai. J Biochem, 120: 335–345.
Ohgi, K, Horiuchi, H, Watanabe, H, Iwama, M, Takagi, M, Irie, M (1992). Evidence that three histidine residues of a base non-specific and adenylic acid preferential ribonuclease from Rhizopus niveus are involved in the catalytic function. J Biochem, 112: 132–138.
Richman, AD, Broothaerts, W, Kohn, JR (1997). Self-incompatibility RNases from three plant families: homology or convergence? Am J Bot, 84: 912–917.
Richman, AD, Uyenoyama, MK, Kohn, JR (1996). Allelic diversity and gene genealogy at the self-incompatibility locus in the Solanaceae. Science, 273: 1212–1216.
Richman, AD, Kohn, JR (1999). Self-incompatibility alleles from Physalis: implications for historical inference from balanced genetic polymorphisms. Proc Natl Acad Sci USA, 96: 168–172.
Rozas, J, Rozas, R (1997). DnaSP version 2.0: a novel software package for extensive molecular population genetics analysis. Comp Appl Biosci (CABIOS), 13: 307–311.
Saba-el-Leil, M, Rivard, S, Morse, D, Cappadocia, M (1994). The S11 and S13 self incompatibility alleles in Solanum chacoense Bitt. are remarkably similar. Plant Mol Biol, 24: 571–583.
Sassa, H, Nishio, T, Kowyama, Y, Hirano, H, Koba, T, Ikehashi, H (1996). Self-incompatibility (S) alleles of the Rosaceae encode members of a distinct class of the T2/S ribonuclease superfamily. Mol Gen Genet, 250: 547–557.
Tanaka, T, Nei, M (1989). Positive darwinian selection observed at the variable-region genes of immunoglobulins. Mol Biol Evol, 6: 447–459.
Tao, R, Yamane, H, Sugiura, A (1999). Cloning of genomic DNA sequences encoding S1, S3, and S6-RNases (Accession Nos. AB03185, AB03186, AB03187 and AB01388) from sweet cherry (Prunus avium L.). Plant Physiol 121: 1057
Tseng, H (1938). Self-sterility in Antirrhinum and Petunia. J Genet, 36: 127–138.
Thompson, J, Gibson, TJ, Plewniak, F, Jeanmougin, F, Higgins, DG (1997). The ClustalX window interface: flexible stategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res, 25: 4876–4882.
Ushijima, K, Sassa, H, Tao, R, Yamane, H, Dandekar, AM, Gradziel, TM et al (1998). Cloning and characterization of cDNAs encoding S-RNases from almond (Prunus dulcis): primary structural features and sequence diversity of the S-RNases in Rosaceae. Mol Gen Genet, 260: 261–268.
Vieira, CP, Vieira, J, Charlesworth, D (1999). Evolution of the cycloidea gene family in Antirrhinum and Misopates. Mol Biol Evol, 16: 1474–1483.
Wang, X, Hughes, AL, Tsukamoto, T, Ando, T, Kao, TH (2001). Evidence that intragenic recombination contributes to allelic diversity of the S-RNase gene at the self-incompatibility (S) locus in Petunia inflata. Plant Physiol, 125: 1012–1022.
Wright, S (1939). The distribution of self-sterility alleles in populations. Genetics, 24: 538–552.
Xue, Y, Carpenter, R, Dickinson, HG, Coen, ES (1996). Origin of allelic diversity in Antirrhinum S locus RNase. Plant Cell, 8: 805–814.
Ye, ZH, Droste, DL (1996). Isolation and characterization of cDNAs encoding xylogenesis-associated and wounding-induced ribonucleases in Zinnia elegans. Plant Mol Biol, 30: 697–709.
Zurek, DM, Mou, B, Beecher, B, McClure, B (1997). Exchanging sequence domains between S-RNases from Nicotiana alata disrupts pollen recognition. Plant J, 11: 797–808.
Acknowledgements
We thank Jorge Vieira for helpful comments on the manuscript. CPV was supported by the Commission of the European Communities (Grant ERBFMBICT 972455), and DC by an NERC Senior Research Fellowship.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Vieira, C., Charlesworth, D. Molecular variation at the self-incompatibility locus in natural populations of the genera Antirrhinum and Misopates. Heredity 88, 172–181 (2002). https://doi.org/10.1038/sj.hdy.6800024
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.hdy.6800024
Keywords
This article is cited by
-
Genetics Behind Sexual Incompatibility in Plants: How Much We Know and What More to Uncover?
Journal of Plant Growth Regulation (2023)
-
Inferences on specificity recognition at the Malus×domestica gametophytic self-incompatibility system
Scientific Reports (2018)
-
No evidence for Fabaceae Gametophytic self-incompatibility being determined by Rosaceae, Solanaceae, and Plantaginaceae S-RNase lineage genes
BMC Plant Biology (2015)
-
Different Positively Selected Sites at the Gametophytic Self-Incompatibility Pistil S-RNase Gene in the Solanaceae and Rosaceae (Prunus, Pyrus, and Malus)
Journal of Molecular Evolution (2007)
-
Allelic diversity of S-RNase at the self-incompatibility locus in natural flowering cherry populations (Prunus lannesiana var. speciosa)
Heredity (2004)
