
Abstract
Purpose
We reviewed all children presenting with microcephaly and bilateral congenital cataract in our paediatric cataract clinic, to identify their underlying diagnosis and clinical features that could help in early diagnosis of such conditions.
Methods
We screened our cataract database to collect data on all children presenting to our institute with cataract before 1 year of age for a period of 5 years (1997–2001) inclusive. We found 166 cases of cataract, of which 85 were bilateral. Of the 85 children with bilateral cataract, five also had microcephaly. The case notes of these five children were retrospectively reviewed for age at presentation, ocular and systemic examination findings, results of electro diagnostic testing, outcome of cataract surgery, and any additional investigation results.
Results
In our series, three children were diagnosed with early-onset Cockayne syndrome (CS2), one was diagnosed with Micro syndrome and one with Hallermann–Streiff syndrome. Electrodiagnostic testing was abnormal in four of the five cases, and growth failure was present in all five. There was a good outcome from surgery in the three children with CS2 and the child with Hallermann–Strieff. There was a poor outcome in the child with Micro syndrome.
Conclusions
The presence of microcephaly in children presenting with bilateral cataracts in infancy is strongly suggestive of a syndromic cause. Early-onset Cockayne syndrome was the commonest underlying diagnosis in our series, but clinicians should be aware of other possibilities, and we discuss the differential diagnosis. Head circumference should be checked routinely in children with congenital cataract.
Similar content being viewed by others
Introduction
Childhood cataracts are a major cause of visual impairment and blindness throughout the world,1 and represent an important problem in paediatric ophthalmology. It has been estimated that almost three children of every 10 000 born in the United Kingdom each year will have congenital or infantile cataracts.2 In two-thirds of these children, the cataracts are bilateral. One further child in every 10 000 will have cataract diagnosed by the age of 15 years. Cataract has also been reported as the commonest congenital ocular malformation, with a prevalence of 2.7 per 10 000 births.3 The most common aetiologies include intrauterine infections, metabolic disorders, and genetically determined syndromes. Primary prevention is possible for infectious causes through immunisation programmes, but regardless of cause, early diagnosis and prompt treatment are necessary to minimise the visual sequelae.
The aim of this study was to review a subgroup of children with congenital cataract and microcephaly presenting in our paediatric cataract clinic, to identify clinical features that could help in early diagnosis of the underlying syndrome, and to review the outcome of cataract surgery and determine the visual prognosis.
Materials and methods
We screened our database for all children presenting to our hospital with cataract at age less than 1 year, over a 5-year period (1997–2001 inclusive). Of 166 cases of cataract, 85 were bilateral. Of these, five children also had microcephaly, defined as a head circumference below two standard deviations from the mean for age and sex. Their clinical notes were retrospectively reviewed for birth history, age of presentation with cataract, ocular features, results of electrodiagnostic test, systemic features, the results of any other investigations, and any underlying syndromic diagnosis.
Case 1
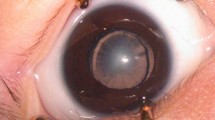
This boy was born to healthy unrelated parents at term by emergency Caesarean section for foetal distress weighing 2.78 kg. He presented with dense white cataracts at the age of 6 weeks. He had horizontal nystagmus. His corneas were clear with well dilating pupils. He was investigated for Lowes syndrome, chromosomal analysis and galactokinase level and was found to be normal. Cataract surgery was performed at the age of 7 weeks in both the eyes a few days apart. This was followed by correction of his aphakia with contact lenses. He had a pigmentary retinopathy and hypoplastic optic discs in both eyes. The ERG amplitudes were below the 5th percentile of normal range. The flash VEP was normal, but although present to moderate check sizes, the pattern VEP to 100′ was increased in latency. He developed dry eyes (he does not cry tears). Despite feeding well, he failed to put on weight. At his further follow-up at the age of 4 months, he was noted to be microcephalic, head circumference 34.5 cm (below 0.4th percentile), length 55.7 cm (below 0.4th percentile) and weight 4.19 kg (below 0.4th percentile), and to have deep-set eyes. He had also developed facial erythema resembling sunburn following minimal exposure to sun (Figure 1). A skin biopsy was performed, which supported the diagnosis of early-onset Cockayne syndrome. Following surgery he was visually attentive and fixed and followed large bright objects and soap bubbles well. His subsequent growth and developmental progress have been very poor, although he is a generally contented child.

Photograph showing deep-set eyes sunburn after minimal exposure to sunlight.
Case 2
This girl was born to healthy unrelated parents at term by normal delivery. Intra-uterine growth retardation was noted from 32 weeks gestation. Her birth weight was 2.36 kg (0.4th percentile) and head circumference was 32.4 cm (0.4th–2nd centile). She was noted to be mildly hypertonic and her cranial ultrasound showed mild ventricular dilatation. She failed her hearing test at birth and had delayed auditory potentials. Bilateral cataracts were detected at 6 weeks of age. The cataracts were mild, with dots and posterior subcapsular lens opacities. She also had poorly dilating pupils. At 9 weeks and 24 months of age, her mixed rod cone ERGs were of subnormal amplitude. Pattern-reversal VEPs were of small amplitude, but evident to large and moderate checks at normal latency for age (latencies maturing appropriately at 24 m, but amplitudes remained small). No surgery was performed as the cataracts were thought to be not visually significant and she was kept under review. She fed poorly and failed to thrive. At 13 weeks of age, her weight was 3.6 kg (below 0.4th percentile), length 52 cm (below 0.4th percentile), and head circumference was 36 cm (below 0.4th percentile). Her mother also reported an episode of skin hypersensitivity to sunlight. At her paediatric review at 4 months, she was found to have global developmental delay. Her TORCH screen, urinary reducing substances, U&E, thyroid function tests, and chromosomal analysis were found to be normal. An MRI brain scan showed no structural abnormality, and myelination was found to be normal. Skin fibroblasts were sent for studies of DNA repair and supported a diagnosis of early-onset Cockayne syndrome.
At the age of 1 year, the cataract in her left eye became visually significant and she had left lens aspiration performed with intraocular lens implantation. Her fundus examination was normal. She was visually attentive and fixed and followed well with both her phakic and pseudophakic eyes. She continued to show marked growth retardation, gastro-oesophageal reflux, slow feeding for which a gastrostomy tube was placed, and severe developmental delay with hypertonicity. At 4 years of age she was able to sit unsupported and stand with support, but had no words. She was interactive and contented. She subsequently developed a kyphoscoliosis. She died peacefully age 7 years 5 months.
Case 3
This girl was born at term by normal delivery to healthy unrelated parents weighing 2.66 kg (2nd–9th centile). The mother was exposed to Rubella during pregnancy but was rubella immune. The child had evidence of slowing of head growth on scans from 30 weeks gestation. She had dense white cataract with small pupils detected at 6-week check. Her mother reported an episode of facial skin peeling resembling sunburn following brief sun exposure. Cataract surgery was performed at 6 weeks of age in both eyes. The right fundus was normal and the left fundus showed pigmentary retinopathy. ERG was subnormal to predominantly rod-driven stimuli at age 7 months, and showed signs of deterioration by 15 months. Flash VEPs were within normal limits, but pattern VEPs were poor. Her head circumference was 32.5 cm at 5 weeks (below 0.4th percentile) and her growth parameters progressively fell away from the centiles. She was found to have a moderate bilateral 60 dB hearing loss, for which she wore aids bilaterally. She was investigated for TORCH, urinary reducing substances, long-chain fatty acid, galactokinase, skeletal survey, metabolic screen, and chromosomal analysis, and all was found to be normal. She also had an MRI of the brain performed, which showed polymicrogyria of right fronto-temporal region compatible with migration defect disorder. A skin biopsy was performed at 7 weeks of age, which supported the diagnosis of early-onset Cockayne syndrome. She was subsequently found to have compound heterozygous mutations in the CSB gene. At present she exhibits good visual behaviour with aphakic glasses. By 4 years of age, she had a length of 66.8 cm, weight 4.61 kg, and head circumference of 35.1 cm (all far below the 0.4th centile). She had never learned to sit unsupported, but was able to roll. She had acquired three short words. She became progressively weaker and died at the age of 4 years 4 months.
Case 4
This baby girl was born 11 days prematurely by normal delivery. There was no family history of consanguinity. She had bilateral dense cataract at birth and poorly dilating pupils and microphthalmia. She had bilateral cataract surgery at age of 3 weeks, 3 days apart. It was difficult to see the fundus due to poorly dilating pupils. She had poor visual outcome following surgery and was occasionally fixing and not following. At 27 months, her mixed rod cone ERGs were of subnormal amplitude and flash and pattern VEPs were markedly degraded, suggesting poor acuity levels. At the age of 2 months, paediatricians were concerned about poor head growth and general development. At the age of 11, months her head circumference was 40 cm (below 0.4th percentile). She developed severe global developmental delay. Her hearing test was within normal limits. She had feeding difficulties and developed spastic quadriplegia. Her TORCH screen, urinary amino acid, urine organic acid, urinary reducing substance, and chromosomal analyses were normal. She had an MRI performed, which showed periventricular leukomalacia. At the age of 3 years, her length was 84 cm (below 0.4th percentile), weight was 9.8 kg (below 0.4th percentile), and head circumference was 43.7 cm (below 0.4th percentile). Her skin biopsy did not support the diagnosis of Cockayne syndrome. Later she was diagnosed with Micro syndrome.
Case 5
This 4-week-old boy was referred to eye clinic with bilateral congenital cataract. He was born at full term by elective caesarean section, weighing 2.86 kg, to an HIV-positive mother who had received antiretroviral treatment. PCR testing for HIV at 6 days and 4 weeks confirmed that he was not infected. In addition to the cataracts, he was noted to have micrognathia and pinched nares. His mother reported that the child had noisy breathing since childhood. On ocular examination he had roving eye movements, bilateral microphthalmos, blue sclera, and bilateral dense cataract. The ultrasound scan showed no posterior hyperplastic primary vitreous or retinal detachment. At age 1 month, his mixed rod cone ERG was smaller than the average for young age to the brightest flash, but a consistent flash VEP at normal latency for young age was evident. He had bilateral lensectomies performed at 5 weeks of age, 2 days apart. His cardiac examination and echocardiogram was within normal limits. As he grew older there was concern regarding his poor growth. At the age of 8 months when reviewed by the paediatric team, his weight was 6.8 kg (below 3rd centile). His head circumference was 46 cm (0.4th percentile) and his length 72 cm (below 0.4th percentile). He had global developmental delay. He failed language, fine motor, and personal social milestones at the 9-month level. At the age of 2 years, he developed upper airway obstruction, which required tracheostomy and later tracheo-cutaneous fistula. A diagnosis of Hallermann–Streiff syndrome was made. Due to narrow palpebral fissures, contact lens fitting was found difficult and he was advised aphakic glasses. At his last ophthalmic follow-up at the age of 5 years he was mainly fixing with his left eye and had developed variable right convergent squint.
Results
In our series, three children were diagnosed with early-onset Cockayne syndrome, one with Micro syndrome, and one with Hallermann–Streiff syndrome. All five children had growth failure as well as microcephaly (Figure 2, Figure 3). A history of skin photosensitivity was strongly suggestive of Cockayne syndrome. Two out of the three children with Cockayne syndrome also had hearing loss, pigmentary retinopathy, and poorly dilating pupils. The electroretinogram was below the normal range in all the cases (Figure 4), in one case (3) showing a deterioration.

Female cases showing microcephaly and growth failure (X=case 2, •=case 3, ▪=case 4). © Child Foundation Trust.

Male cases showing microcephaly and growth failure (•=case 1, ▪=case 5). © Child Foundation Trust.

The 95th and 5th percentile ranges of ERG b-wave amplitudes elicited to different stimuli. Patient data are numbered and illustrate how they fall just outside the lower limit of normal, particularly to predominantly rod-driven stimuli.
Discussion
The purpose of this study was to review the children seen by us, who had microcephaly in addition to bilateral congenital or infantile-onset cataracts. In our experience, there is a strong likelihood of an underlying syndrome diagnosis in this group. The diagnoses were important to make because of their implications for longer-term prognosis, and because many have an underlying autosomal recessive basis with 25% recurrence risks for future pregnancies of the parents. The most frequent diagnosis in our series was early-onset Cockayne syndrome.
Cockayne syndrome is an autosomal recessive disorder first described in 19364 as an association of dwarfism, retinal atrophy, and deafness in two siblings. Classical Cockayne syndrome (CS1) usually presents between 4–5 years of age, and is characterised by arrest of development in a previously normal child associated with progressive facial changes, particularly a sunken appearance around the eyes. This is followed by progressive mental and physical retardation, a prematurely aged appearance, microcephaly, ataxia, and sensorineural deafness.5 Many have cutaneous photosensitivity and a history of redness or skin peeling following sun exposure should be sought. Progressive neurological deterioration leads to death by the second to third decade of life. Reported ocular findings include dry eyes, miotic pupils, pigmentary retinopathy, optic atrophy, facial palsy, cataract, corneal opacities, nystagmus, and microcornea.6 Early-onset Cockayne syndrome (CS2) presents at birth or in infancy with similar facial and somatic features, but in a more rapidly progressive form, death usually occurring earlier by the age of 6 or 7 years. CS2 is probably the same as cerebro-oculo-facio-skeletal syndrome (COFS) and CAMFAK. The diagnosis of both types of Cockayne syndrome requires a skin biopsy to study fibroblast response to UV light. Characteristically, the fibroblasts show defective recovery of RNA synthesis following exposure to ultraviolet-C light, because of a defect in the transcription-coupled nucleotide excision repair mechanism responsible for removing photoproducts from active genes.7, 8 No abnormalities of DNA repair or chromosome breakage are found in lymphocytes. Cockayne syndrome is genetically heterogeneous. At a cellular level, there are two main biochemical complementation groups known as CSA and CSB. CSA results from mutations in the ERCC8 (excision repair cross-complementing group 8) gene, whereas CSB results from mutations in ERCC6. There are additional patients with overlapping features of Cockayne syndrome and xeroderma pigmentosa who have mutations in one of three xeroderma pigmentosa genes, XPB, XPD, or XPG.9 Mutation screening is not routinely available. Prenatal diagnosis is possible from 11 weeks gestation by studying the response of cultured chorionic villus cells or amniocytes to UV light.
The cataracts associated with Cockayne syndrome may be zonular, polar, or punctate. Cortical posterior vacuoles and nuclear cataracts have also been described.10 We also did not find a specific type of cataract (Figure 5). The outcome of cataract surgery in children with CS2 has not been reported previously to our knowledge. All three of our CS2 patients had cataract surgery performed at an early age, with good visual behaviour following surgery. Despite evidence of pigmentary retinopathy (cases 1 and 3), we believe that operating on these children particularly early on, optimises their visual potential.

Dense nuclear sclerosis and posterior lenticonus in a CS2 patient.
Micro syndrome was first reported by Warburg et al11 as an autosomal recessive condition characterised by microcornea, cataract, optic atrophy, absent VEP with less impact on ERG, microcephaly, severe mental retardation, hypotonic spastic diplegia, cortical gyral abnormalities, hypoplasia of corpus callosum, and hypothalamic hypogenitalism. Ainsworth et al12 emphasised the distinctive eye findings of microphthalmos, microphakia, characteristic lens opacity, small non-dilating atonic pupils with minimum vision due to severe cortical blindness. Micro differs from CS2 by manifesting less prenatal and postnatal growth deficiency, poor visual perception even after early cataract extraction, normal fibroblast DNA repair studies, cortical dysplasia rather than progressive brain atrophy on MRI scan, and a better prognosis for longer-term survival.13 Our case 4 was similar to that described in the literature, with microcephaly, microphthalmos, congenital cataract, poor vision, normal hearing, global developmental delay, spastic quadriplegia, and normal skin biopsy. Around two-thirds of patients with Micro syndrome have mutations in the gene encoding catalytic subunit of RAB3GAP (RAB3 GTP-ase-activating protein), known as RAB3GAP1.14 RAB3GAP acts as a regulator of calcium-mediated hormone and neurotransmitter exocytosis. In the remainder, the gene is currently unknown.
Hallermann–Streiff syndrome is a sporadic condition for which the cause is currently unknown. It is usually recognisable at birth and typically presents with congenital cataracts, often with bilateral microphthalmia. The head shape is brachycephalic, with parietal bossing, wide fontanelles, and wide sutures with Wormian bones. There is micrognathia and malar hypoplasia, a thin nose with atrophic overlying skin, hypodontia, and hypotrichosis.15 Low birth weight is common with proportionate postnatal growth retardation in the remainder. The upper airways are narrow and contribute to frequent respiratory complications early on. Early feeding problems are also common. Adult stature is short, but intelligence is usually normal.
There are a number of other closely overlapping syndromes, which should also be considered in infants with cataracts and microcephaly, as summarised in Table 1.
Martsolf syndrome is also associated with cataracts, short stature, hypogonadism, and severe mental retardation. It appears to be autosomal recessive.16 There are overlaps with Micro syndrome, and recently a homozygous missense mutation in RAB3GAP2, the non-catalytic subunit of RAB3GAP, has been reported in one family, providing a likely molecular basis for the clinical overlap.17
Marinesco–Sjogren syndrome is an autosomal recessive multisystem disorder characterised by cataracts, cerebellar hypoplasia, growth retardation, mild–moderate mental retardation, hypotonia, demyelinating neuropathy, seizures, hypogonadism, and skeletal abnormalities including kyphoscosiosis, contractures, and pes planus.18 In some families it results from mutations in a gene called SIL1 encoding an endoplasmic reticulum glycoprotein BAP (BIP-associated protein). Additional reported eye findings include optic atrophy, glaucoma, microphthalmia, and retinal degeneration.19
Congenital cataract–facial dysmorphism–neuropathy (CCFDN) syndrome is an autosomal recessive syndrome described to date only in Vlax Roma gypsies. It resembles Marinesco–Sjogren syndrome, but is now known to result from mutations in a different gene, CTDP1, encoding a phosphatase subunit required for the regulation of RNA polymerase II.20
Smith–Lemli–Opitz syndrome is another autosomal recessive cause of infantile cataracts and microcephaly, which is usually suspected soon after birth, because of associated malformations including cleft palate, ptosis, ambiguous genitalia in males, polydactyly, and syndactyly, particularly between the second and third toes, and malformations of the brain and kidneys.21 The diagnosis is confirmed by finding high levels of 7-dehydrocholesterol, an intermediary in the biosynthesis of cholesterol. The condition results from mutations in the 7-dehydrocholesterol reductase gene, and prenatal testing is available using biochemical or molecular genetic methods.
Disorders of peroxisome metabolism are not usually associated with early microcephaly, but may be associated with hypotonia, skeletal abnormalities, hepatosplenomegaly, brain, and renal abnormalities in addition to cataracts. They can be screened by using X-ray for epiphyseal stippling, testing for abnormalities of very long-chain fatty acids (VLCFAs), and urine screening for organic acids. Confirmation of the underlying enzyme defect in cultured fibroblasts is important for prenatal testing in future pregnancies.
Neu–Laxova syndrome is a further autosomal recessive cause of congenital cataracts and microcephaly, but is easily recognised by the sloping forehead, micrognathia, protruding eyes often with absent lids, ichthyosis and absent hair, contractures, and syndactyly. Survival beyond the first week is rare. The causative gene is not known.
Determining the aetiology of congenital or infantile-onset cataract is important for predicting both visual and overall prognoses, and for counselling parents as to recurrence risks. A number of factors should be established, including the type of cataract, whether they are bilateral, and whether they are associated with other abnormalities of the eye such as microphthalmia, anterior chamber malformation, or abnormalities of the optic nerve or retina. Visual electrophysiology is a useful tool in assessment of these children and if available, should be performed to help in diagnosis. A thorough physical examination for evidence of other malformations or anomalies should be performed, and that should include documentation of growth and head circumference. Microcephaly or growth failure may not be evident at birth, hence regular examination by a paediatrician for such cases for the first 2 years to detect early signs of systemic association should be performed. A family and pregnancy history should be taken, looking in particular for evidence of familial cataracts, congenital infection or other teratogenic exposure, and consanguinity. The parents' eyes should be examined for lens opacity.
Routine investigations should include screening for evidence of congenital infections, urine testing for reducing substances, protein, amino acids and organic acids, and screening for galactokinase deficiency. In those with additional abnormalities, further testing should be considered, in particular, karyotyping for chromosome imbalance, metabolic screening for disorders of peroxisome or cholesterol metabolism, and referral for a clinical genetics opinion. Further investigations will depend on the examination findings, family history, and the child's subsequent developmental progress. A proportion of cases with cataract and microcephaly will not categorise into a recognised syndrome, hence monitoring for growth, head circumference, or systemic features is important, and features of Cockayne syndrome and Micro syndrome in particular should be specifically sought. Poor VEPs to flash stimulation as well as pattern and poor visual outcome despite early cataract surgery should raise the suspicion of Micro syndrome.
References
Thylefors B . A global initiative for the elimination of avoidable blindness. Indian J Ophthalmol 1998; 46 (3): 129–130.
Rahi JS, Dezateaux C . Measuring and interpreting the incidence of congenital ocular anomalies: lessons from a national study of congenital cataract in the UK. Invest Ophthalmol Vis Sci 2001; 42 (7): 1444–1448.
Stoll C, Alembik Y, Dott B, Roth MP . Congenital eye malformations in 212,479 consecutive births. Ann Genet 1997; 40 (2): 122–128.
Cockayne EA . Dwarfism with retinal atrophy and deafness. Arch Dis Child 1936; 11: 1–8.
Nance MA, Berry SA . Cockayne syndrome: review of 140 cases. Am J Med Genet 1992; 42 (1): 68–84.
Traboulsi EI, De Becker I, Maumenee IH . Ocular findings in Cockayne syndrome. Am J Ophthalmol 1992; 114 (5): 579–583.
Tanaka K, Kawai K, Kumahara Y, Ikenaga M, Okada Y . Genetic complementation groups in Cockayne syndrome. Somatic Cell Genet 1981; 7 (4): 445–455.
Rainbow AJ, Howes M . A deficiency in the repair of UV and gamma-ray damaged DNA in fibroblasts from Cockayne's syndrome. Mutat Res 1982; 93 (1): 235–247.
Spivak G . The many faces of Cockayne syndrome. Proc Natl Acad Sci USA 2004; 101 (43): 15273–15274.
Ferreira RC, Roeder ER, Bateman JB . Cataract in early onset and classic Cockayne syndrome. Ophthalmic Genet 1997; 18 (4): 193–197.
Warburg M, Sjo O, Fledelius HC, Pedersen SA . Autosomal recessive microcephaly, microcornea, congenital cataract, mental retardation, optic atrophy, and hypogenitalism. Micro syndrome. Am J Dis Child 1993; 147 (12): 1309–1312.
Ainsworth JR, Morton JE, Good P, Woods CG, George ND, Shield JP et al. Micro syndrome in Muslim Pakistan children. Ophthalmology 2001; 108 (3): 491–497.
Graham Jr JM, Hennekam R, Dobyns WB, Roeder E, Busch D . MICRO syndrome: an entity distinct from COFS syndrome. Am J Med Genet A 2004; 128 (3): 235–245.
Aligianis IA, Johnson CA, Gissen P, Chen D, Hampshire D, Hoffmann K et al. Mutations of the catalytic subunit of RAB3GAP cause Warburg Micro syndrome. Nat Genet 2005; 37 (3): 221–223.
Cohen Jr MM . Hallermann–Streiff syndrome: a review. Am J Med Genet 1991; 41 (4): 488–499.
Hennekam RC, van de Meeberg AG, van Doorne JM, Dijkstra PF, Bijlsma JB . Martsolf syndrome in a brother and sister: clinical features and pattern of inheritance. Eur J Pediatr 1988; 147 (5): 539–543.
Aligianis IA, Morgan NV, Mione M, Johnson CA, Rosser E, Hennekam RC et al. Mutation in Rab3 GTPase-activating protein (RAB3GAP) noncatalytic subunit in a kindred with Martsolf syndrome. Am J Hum Genet 2006; 78 (4): 702–707.
Slavotinek A, Goldman J, Weisiger K, Kostiner D, Golabi M, Packman S et al. Marinesco–Sjogren syndrome in a male with mild dysmorphism. Am J Med Genet A 2005; 133 (2): 197–201.
Senderek J, Krieger M, Stendel C, Bergmann C, Moser M, Breitbach-Faller N et al. Mutations in SIL1 cause Marinesco–Sjogren syndrome, a cerebellar ataxia with cataract and myopathy. Nat Genet 2005; 37 (12): 1312–1314.
Varon R, Gooding R, Steglich C, Marns L, Tang H, Angelicheva D et al. Partial deficiency of the C-terminal-domain phosphatase of RNA polymerase II is associated with congenital cataracts facial dysmorphism neuropathy syndrome. Nat Genet 2003; 35 (2): 185–189.
Nowaczyk MJ, Waye JS . The Smith–Lemli–Opitz syndrome: a novel metabolic way of understanding developmental biology, embryogenesis, and dysmorphology. Clin Genet 2001; 59 (6): 375–386.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Goyal, R., Thompson, D., Timms, C. et al. Review of cases presenting with microcephaly and bilateral congenital cataract in a paediatric cataract clinic. Eye 22, 273–281 (2008). https://doi.org/10.1038/sj.eye.6702958
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.eye.6702958
Keywords
This article is cited by
-
Neuropädiatrische Differenzialdiagnostik der Mikrozephalie im Kindesalter
Medizinische Genetik (2016)
