
Abstract
Aims:
To describe the phenotype of members of a large Caucasian British family affected by autosomal dominant cone–rod dystrophy due to an R838C mutation in the guanylate cyclase 2D (GUCY2D) gene encoding retinal guanylate cyclase-1 (RETGC-1).
Methods:
Retrospective review of 29 patients from four generations of the same family.
Results:
Visual symptoms usually commenced in childhood. Only two patients, aged 14 and 25 years, had visual acuity compatible with driving. Of the 12 patients aged over 40 years, eight (66%) had vision of counting fingers or worse and were eligible for blind registration in the UK. Of the 29 patients, 18 (62%) had myopia greater than 5 D in at least one eye. Most had discernible macular changes on biomicroscopy, which varied from subtle RPE change to gross macular atrophy. All patients who underwent computerised perimetry exhibited a central or paracentral scotoma with normal peripheral field of vision. Of the 21 patients who underwent electrodiagnostic testing, all exhibited decreased cone function, but rod function was normal in 12 (57%) patients.
Conclusion:
We believe this report highlights the importance of phenotype–genotype correlation in cone and cone–rod dystrophies. Increased understanding of the varying phenotypes associated with different genetic mutations allows appropriate counselling of patients. In addition, the phenotypic characterisation of the natural history of these conditions may prove valuable in the future should therapeutic interventions become available.
Similar content being viewed by others


Introduction
Cone dystrophies are a genetically heterogeneous group of disorders characterised by early deterioration of visual acuity and colour vision.1 Although cone dystrophies with normal rod function may occur, in most cases there is a degree of rod impairment, albeit less severe than that of cone function and often not evident until late in the disease. These patients are described as having cone–rod dystrophies.
Cone and cone–rod dystrophy are genetically heterogeneous with dominant, recessive, and X-linked inheritance patterns described. To date, nine loci for autosomal dominant cone and cone–rod dystrophies have been identified: CORD4, RCD1, AIPL1, CRX, GUCA1A, GUCY2D, RIMS, UNC119, and SEMA4A.
In 1997, Kelsell et al2 localised a gene responsible for autosomal dominant cone–rod dystrophy (ADCRD) in a British family (CORD6) to chromosome 17p. Further work revealed mutations in the guanylate cyclase 2D (GUCY2D) gene encoding retinal guanylate cyclase-1 (RETGC-1) to be responsible.3, 4 Analysis of the original CORD6 family and other families with ADCRD identified single or multiple mutations in codons 837, 838, or 839 of the GUCY2D gene.4, 5, 6, 7 The phenotypes of these families appears to vary depending on the specific mutation and the presence or absence of multiple mutations. There is also some phenotype variation both between and within families with the same mutation.
A large Caucasian family of British origin with ADCRD due to an arginine to cysteine missense mutation at amino-acid residue 838 (R838C) has been attending our department for many years. This paper describes the phenotype of 29 affected members of this family.
Materials and methods
Patients
A pedigree of this family identified 35 members with likely cone–rod dystrophy. Of the 35 family members, 29 were current patients in our department or had attended within the last 5 years. The case notes of all 29 were reviewed. Of the 29 patients, nine had undergone molecular genetic investigation.
Clinical and functional investigations
All 29 patients had undergone clinical examination including Snellen visual acuity and dilated retinal examination. A total of 18 patients had undergone colour vision testing using Ishihara pseudo-isochromatic plates and 13 patients static threshold perimetry using the standard 24-2 threshold protocol on the Humphrey Field Analyser. A total of 21 patients had undergone electrodiagnostic investigation.
Genotyping
Nine of the 29 patients and one unaffected family member had undergone molecular genetic investigation.
Haplotype analysis
Microsatellite markers proximal and distal to the RDS gene on 6p (D6S1582, D6S1552, D6S1017, D6S1672, D6S1549), GUCY2D on 17p (D17S1353, D17S960, D17S786, D17S1796), and the CRX gene on 19q13.3 (D19S902, D19S604) were amplified in a mix containing 40 ng genomic DNA, 5 pmol of forward and reverse primers, 2.5 nmol each dNTP, 100 mM Tris-HCl, 500 mM KCl, 15 mM MgCl2, and 0.25 U Amplitaq Gold Taq polymerase (Applied Biosystems). Forward or reverse primers were labelled with 6-FAM or HEX fluorescent dyes. Reaction conditions were initial denaturation of 95°C for 12 min, followed by 10 cycles of 94°C for 30 s, 55°C for 30 s, 72°C for 1 min, and then 30 cycles of 89°C for 30 s, 55°C for 30 s, and 72°C for 1 min, with a final step of 72°C for 7 min. PCR products were separated by polyacryamide gel electrophoresis on an ABI 377 DNA sequencer and analysed using Genescan 3.0 and Genotyper 2.5 software (Applied Biosystems). Primers sequences were obtained from the Genome Database (http://www.gdb.org/). Tails to reduce stutter bands (GTTTCTT) were introduced to the 5′ end of either the forward or reverse primer.
Identification of mutations in the GUCY2D gene
Exon 13 of the human GUCY2D gene was directly amplified from genomic DNA using primers GUCY2D 13F (5′-GTA GAT GAA TGG TGG CAG CG-3′) and GUCY2D 13R (5′-GAT GGC AGG CAG TGA GGT C-3′). Intronic primers were designed based on the genomic sequence of GUCY2D (http://genome.ucsc.edu/). Approximately 200 ng of patient or control genomic DNA was used in a 50 μl reaction mixture containing 20 pmol forward and reverse primers, 10 nmol each of dTTP, dCTP, and dATP, 7.6 nmol dGTP, 2.5 nmol deaza GTP, 2.5 μl DMSO, 50 μmol betaine, 100 mM Tris-HCl, 500 mM KCl, 15 mM MgCl2, and 1.0 U Amplitaq Gold Taq polymerase (Applied Biosystems). After an initial denaturation of 95°C for 12 min, 40 cycles were performed of 95°C for 1 min, 55°C for 1 min, and 72°C for 2 min, with a final extension step of 72°C for 10 min. The PCR products were examined by 2% agarose gel electrophoresis, purified using spin columns (Qiagen, Crawley, England) and directly sequenced using Big Dye terminators on an ABI 377 genetic analyser (ABI, Foster City, USA) and analysed using Sequence Navigator 1.0.1 software (Applied Biosystems).
Results
Clinical and functional investigations
Figure 1 shows the pedigree of this family. There is clearly autosomal dominant inheritance, with no evidence of nonpenetrance. The phenotype was similar in male and female subjects. In all, 34 family members were reported to be affected, and of these 29 had been examined in our department, the remaining five (I.2, II.3, II.6, III.2, and III.5) were either deceased or had moved away from the area. A diagnosis of cone–rod dystrophy had been made on the basis of clinical examination with or without electrodiagnostic testing. The main clinical and electrophysiological features of these patients are summarised in Table 1.

Pedigree of family with autosomal dominant cone dystrophy. Roman numerals indicate generations; solid symbols, affected family members; open symbols, unaffected family members; squares, males; circles, females; slash, deceased.
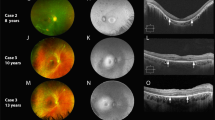
Visual symptoms usually commenced in childhood with 72% of patients experiencing symptoms in the first decade of life and a further 10% in the second decade. Five patients (17%), however, did not notice any problems until the third decade. Only two patients (7%), aged 14 and 25 years, had visual acuity compatible with driving. Of the 12 patients aged over 40 years, eight (66%) had vision of counting fingers or worse and are eligible for blind registration in the UK. Although photophobia was a common symptom, no patients reported problems with nyctalopia. Of the 29 patients, 18 (62%) have myopia greater than 5 D in at least one eye, many of these with myopic astigmatism. A total of 26 patients (90%) had discernible macular changes on biomicroscopy, which varied from subtle RPE change (11 patients) to gross macular atrophy (eight patients). Figure 2 shows a typical fundus appearance in advanced disease with macular atrophy. All patients who underwent computerised perimetry exhibited a central or paracentral scotoma with normal peripheral field of vision. All patients with visual acuity good enough to allow testing with Ishihara pseudo-isochromatic plates were only able to see the test plate. Of the 21 patients who underwent electrodiagnostic testing, all exhibited decreased cone function but rod b-wave electroretinogram (ERG) was of normal latency and amplitude in 12 (57%). Examples of a reduced photopic 30 Hz flicker ERG response are shown in Figure 3.

Fundus photograph of patient III-16 with bilateral macular atrophy.

Examples of photopic 30 Hz flicker electroretinograms. Patient numbers as in Figure 1.
Genotyping
Nine of the 29 patients and one unaffected family member had undergone molecular genetic investigation. Haplotype analysis demonstrated linkage to the GUCY2D gene on chromosome 17 in all nine affected patients, but not the unaffected control. Direct sequencing of the GUCY2D exon 13 in the nine affected family members revealed heterozygosity for a C>T substitution that leads to an arginine to cysteine missense mutation at the amino-acid residue 838 (R838C) (Figure 4). The unaffected family member showed no mutation in exon 13.

A heterozygous C>T mutation results in an arginine to cysteine mutation at codon 838 of the GUCY2D gene.
Discussion
The structural changes of the protein retinal guanylate cyclase (RETGC-1), which arise from mutations in codons 837, 838, and 839 of exon 13 of the GUCY2D gene, are known to cause ADCRD. This protein plays a key role in the phototransduction cascade by controlling intracellular calcium concentration via the influence on cation channels of its enzymatic product, guanosine 3′, 5′-cyclic monophosphate (cGMP). The amino acids affected by these mutations play a role in the dimerisation of the enzyme into its active form. The mutations result in increased stability of the dimer, resulting in raised intracellular calcium concentration in the photoreceptor outer segment.8, 9 It is this high intracellular calcium concentration which is proposed to damage the photoreceptor, resulting in ADCRD.
Recessive mutations in this same gene are responsible for a significant proportion of cases of Lebers Congenital Amaurosis (LCA), a hereditary retinal dystrophy characterised by severe visual impairment from birth.10 Although LCA mutations have been identified in several locations throughout the gene, no case has been localised to exon 13, and it seems likely that the mutations associated with LCA have a more severe effect on protein function than the mutations which result in ADCRD.
In addition, there are differences between the phenotype of families identified to have exon 13 mutations. The original CORD6 family had a missense mutation resulting in the replacement of glutamate by aspartate at codon 837 (E837D) and a mutation at codon 838 which results in the substitution of arginine by serine (R838S).4 Perrault et al5 reported a family with amino-acid substitutions at codons 837, 838, and 839 and there have been descriptions of several single mutations at codon 838 resulting in ADCRD. Although the common feature of these cases is a mutation at codon 838, the families with additional mutations in the adjacent codons do seem to exhibit a severe phenotype with earlier onset and more pronounced rod involvement and these additional changes may have a cumulative effect on protein function.
The family in this report has the R838C mutation. This mutation was first reported by Kelsell et al3 in 1998 as a single mutation resulting in ADCRD in three British families. The same mutation was one of the three mutations in a single family described by Perrault et al5 as discussed above. Although the phenotype of several members of each of the three original British families was described by Downes et al,11 the only previous phenotypic description of a multigenerational family with the R838C mutation was by Van Ghelue et al7 who described the characteristics of 11 members of an affected Norwegian family. The British family in this report appears to exhibit similar characteristics to the Norwegian family. Symptoms tended to begin in the first decade and visual acuity was generally poor, with only two of our patients maintaining visual acuity that met the legal standard for driving. Like the Norwegian family, myopia and astigmatism is common in this family and fundus appearance varies from fine pigment change in the early stages to advanced macular atrophy later in the disease. Unlike the Norwegian report where preserved rod function was limited to one branch of the family, 57% of our family who underwent electrodiagnostic testing exhibited normal rod b-wave ERG. These patients were not restricted to one branch of the family and included patients with very poor visual acuity and absent cone responses on electrodiagnostic testing.7 Rod involvement does not therefore seem to be a predominant feature of ADCRD due to R838C mutations, and this appears to apply to all of the single GUCY2D gene mutations, in contrast to the original CORD6 family and the other reported families with multiple GUCY2D mutations.
Our description of the phenotype of a large caucasian family of British origin with ADCRD due to an R838C mutation in the GUCY2D gene confirms this as a disease with variable phenotype in the younger patient. Although most children affected by this condition will be able to be educated in mainstream schools with appropriate support, few will maintain adequate visual acuity to allow them to drive, and by the age of 40 years most will have very poor visual acuity. Rod dysfunction is not a predominant feature in this family and many had a normal rod ERG.
We believe this report highlights the importance of phenotype–genotype correlation in cone–rod dystrophies. Increased understanding of the varying phenotypes associated with different genetic mutations allows appropriate counselling of patients. In addition, the phenotypic characterisation of the natural history of these conditions may prove valuable in the future, should therapeutic interventions become available.
References
Simunovic MP, Moore AT . The cone dystrophies. Eye 1998; 12: 553–565.
Kelsell RE, Evans K, Gregory CY, Moore AT, Bird AC, Hunt DM . Localisation of a gene for dominant cone–rod dystrophy (CORD6) to chromosome 17p. Hum Mol Genet 1997; 6(4): 597–600.
Kelsell RE, Gregory-Evans K, Payne AM, Perrault I, Kaplan J, Yang RB et al. Mutations in the retinal guanylate cyclase (RETGC-1) gene in dominant cone–rod dystrophy. Hum Mol Genet 1998; 7(7): 1179–1184.
Payne AM, Morris AG, Downes SM, Johnson S, Bird AC, Moore AT et al. Clustering and frequency of mutations in the retinal guanylate cyclase (GUCY2D) gene in patients with dominant cone–rod dystrophies. J Med Genet 2001; 38(9): 611–614.
Perrault I, Rozet JM, Gerber S, Kelsell RE, Souied E, Cabot A et al. A RETGC-1 mutation in autosomal dominant cone-rod dystrophy. Am J Hum Genet 1998; 63(2): 651–654.
Weigell-Weber M, Fokstuen S, Torok B, Niemeyer G, Schinzel A, Hergersberg M . Codons 837 and 838 in the retinal guanylate cyclase gene on chromosome 17p: hot spots for mutations in autosomal dominant cone–rod dystrophy. Arch Ophthalmol 2000; 118(2): 300.
Van Ghelue M, Ericksen HL, Ponjavic V, Fagerhein T, Andreasson S, Forsman-Semb K et al. Autosomal dominant cone–rod dystrophy due to a missense mutation (R838C) in the guanylate cyclase 2D gene (GUCY2D) with preserved rod function in one branch of the family. Ophthal Genet 2000; 21(4): 197–209.
Tucker CL, Woodcock SC, Kelsell RE, Ramamurthy V, Hunt DM, Hurley JB . Biochemical analysis of a dimerization domain mutation in RETGC-1 associated with dominant cone–rod dystrophy. Proc Natl Acad Sci USA 1999; 96(16): 9039–9044.
Wilkie SE, Newbold RJ, Deery E, Walker CE, Stinton I, Ramamurthy V et al. Functional characterization of missense mutations at codon 838 in retinal guanylate cyclase correlates with disease severity in patients with autosomal dominant cone–rod dystrophy. Hum Mol Genet 2000; 9(20): 3065–3073.
Hanein S, Perrault I, Gerber S, Tanguy G, Barbet F, Ducroq D et al. Leber congenital amaurosis: comprehensive survey of the genetic heterogeneity, refinement of the clinical definition, and genotype–phenotype correlations as a strategy for molecular diagnosis. Hum Mutat 2004; 23(4): 306–317.
Downes SM, Payne AM, Kelsell RE, Fitzke FW, Holder GE, Hunt DM et al. Autosomal dominant cone–rod dystrophy with mutations in the guanylate cyclase 2D gene encoding retinal guanylate cyclase-1. Arch Ophthalmol 2001; 119(11): 1667–1673.
Acknowledgements
We thank Torbay Medical Research Fund for sponsoring the molecular analysis.
Author information
Authors and Affiliations
Corresponding author
Additional information
Proprietary interests: Nil
Rights and permissions
About this article
Cite this article
Smith, M., Whittock, N., Searle, A. et al. Phenotype of autosomal dominant cone–rod dystrophy due to the R838C mutation of the GUCY2D gene encoding retinal guanylate cyclase-1. Eye 21, 1220–1225 (2007). https://doi.org/10.1038/sj.eye.6702612
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.eye.6702612
Keywords
This article is cited by
-
Phenotypic characterization of autosomal dominant progressive cone dystrophies associated with a heterozygous variant c.2512C>T of GUCY2D gene in a large kindred
Eye (2022)
-
Longitudinal OCT and OCTA monitoring reveals accelerated regression of hyaloid vessels in retinal degeneration 10 (rd10) mice
Scientific Reports (2019)
-
GUCY2D mutations in a Chinese cohort with autosomal dominant cone or cone–rod dystrophies
Documenta Ophthalmologica (2015)
-
A detailed phenotypic description of autosomal dominant cone dystrophy due to a de novo mutation in the GUCY2D gene
Eye (2014)
-
Clinical course of cone dystrophy caused by mutations in the RPGR gene
Graefe's Archive for Clinical and Experimental Ophthalmology (2011)
