
Abstract
Aims
Gelatinous drop-like corneal dystrophy (GDLD) is an early-onset, autosomal recessive condition characterised by amyloid deposits within the cornea. We report the histopathological and molecular genetic findings in a Caucasian child with GDLD who also exhibited global developmental delay.
Methods
Bilateral lamellar keratoplasty was carried out at age 6 and 7 years. Tissue was fixed for light and electron microscopy, including immunoelectronmicroscopy. The coding region of the M1S1 gene was screened for mutations in the affected proband and available relatives, using DNA extracted from mouthwashes.
Results
Nodular deposits, which were present subepithelially and in the central superficial stroma, stained typically for amyloid with PAS and Congo red. A nodular deposit of amyloid, together with large amounts of lactoferrin and sparse amounts of keratoepithelin (βig-h3), was present in the central superficial stroma, causing destruction of Bowman's layer and elevation of the thinned, degenerate epithelium. Around the deposit zone, the stroma exhibited large numbers of thick filamentous proteoglycan deposits. While the affected child was homozygous for a novel A1133 C single-nucleotide polymorphism (SNP) that resulted in an aspartic acid to alanine substitution at position 173 of the M1S1 coding sequence, this polymorphism was also found at relatively high frequency in a sample of normal controls, enabling exclusion of the M1S1 gene as the disease locus.
Conclusion
Increased epithelial permeability in GDLD may be explained in part by an altered membrane permeability of the superficial epithelial cells. An association with developmental delay has not been reported previously.
Similar content being viewed by others

Introduction
Gelatinous drop-like corneal dystrophy (GDLD) (OMIM #204870) is an early-onset, autosomal recessive corneal dystrophy, reported globally1, 2, 3 but most commonly in Japan.4, 5 It presents in the first decade of life with bilateral, axial, elevated, mulberry-like or gelatinous lesions, due to amyloid deposition in the superficial cornea. With time, the cornea vascularises and the deposits spread laterally and deeply within the stroma, leading to a progressive loss in vision. However, there is considerable phenotypic variation.6 Penetrating keratoplasty and phototherapeutic keratoplasty (PTK) is followed by early recurrence.7 Some success with limbal stem cell transplantation suggests an important epithelial contribution.8 The disorder has been mapped to chromosome 1p and associated, in most, but not all cases with mutations in the M1S1 gene.9, 10, 11, 12, 13, 14, 15, 16, 17 Yoshida et al18 reported that the Q118X mutation of the M1S1 gene could produce either a gelatinous drop-like region or band-shaped opacities.
We report here the clinicopathological features and molecular genetic analysis of a child who presented with the phenotypic features of GDLD in infancy and underwent bilateral, lamellar keratoplasties. We were particularly interested to study the expression of lactoferrin and keratoepithelin. Lactoferrin is present in high amounts in corneas with GDLD but absent from normal corneas.19 Keratoepithelin is an extracellular matrix protein (also referred to as TGF beta-induced protein, or βig-h3) which is incorporated into deposits in three major forms of dominant corneal dystrophy caused by mutations in the BIGH3 gene, that is, lattice dystrophy (types I, III, and IV), granular, ‘Avellino’, and Thiel Behnke dystrophies.20 We also screened the coding region of the M1S1 gene for mutations.
Methods
Clinical findings
The affected proband was a Caucasian male, born of a normal pregnancy, whose development was noted to be delayed at age 6 weeks. At the age of 14 months, he was referred with a history of possible squint, along with the presence of axial, elevated spots on each cornea (Figure 1a). At EUA the lesions were noted to stain markedly with fluorescein. Uncorrected vision fluctuated between 6/24 and 6/60. A right, followed by a left lamellar keratoplasty, was performed at the ages of 6 and 7 years, respectively (7.75 into 7.5 mm). At EUA, both corneal grafts were clear on 27 July 1999, apart from a mild intrastromal granularity on the right. At the time of writing, clinical examination at the slit lamp shows the presence of early, superficial recurrence of the disorder in each cornea. At the age of 3.5 years, language development was said to have been delayed to about 2 years, and at the age of 6 year his educational level was assessed as 4 years. He manages well at a sighted school.

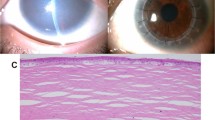
(a) Clinical photograph of GDLD eye showing the superficial, gelatinous, axial deposit of a 7-year-old child. (b) Light micrograph of GDLD cornea showing degenerate epithelium (E) and nodular deposits (AM). (c) Light micrograph of GDLD cornea showing thick epithelium (E) and fibrous pannus (P). (d) GDLD cornea stained with Congo red showing nodular deposits of amyloid (AM).
Histology and immunoelectron microscopy
Corneal tissue was divided and fixed immediately in either 3% formalin, 3% glutaraldehyde, and 1% osmium tetroxide in 0.1 M. phosphate buffer, 3% glutaraldehyde containing 0.05% cuprolinic blue, or 4% paraformaldehyde, for light microscopy, electron microscopy, proteoglycan localisation, or immunoelectron microscopy, respectively.21
For immunoelectron microscopy, tissue was embedded at low temperature as described.21 Rabbit polyclonal anti-human keratoepithelin (gift from Dr Kelly Benett, Bristol Myers Squibb, USA), and rabbit antilactoferrin were visualised with 10 nm immunogold-conjugated anti-rabbit (Biocell, Cardiff, UK).
Molecular genetics
Genomic DNA was extracted from saline mouthwashes, essentially as described.22 The single exon of the M1S1 gene (GenBank NM002353) was amplified using 3 sets of PCR primers to generate overlapping products, which were screened for mutations by direct sequencing. The primers used were GDLD–3F and GDLD–5R17 (681 bp), 5′ GGA TGT GTC ACC CAA ATA CCA and 5′ CTT GAG CAG CAG ACA CTT GGA (423 bp), and 5′ CCT ACT ACT TCG AGA GGG ACA and 5′ CAG GAA GCG TGA CTC ACT T (382 bp). The frequency of the novel A1133 C polymorphism was analysed in 280 chromosomes from control subjects (approximately 80% Caucasians, 20% Asians) by PCR with primers GDLD–3F and GDLD–5R followed by restriction digestion with Eae–I (New England Biolabs), which recognises the sequence Y^GGCCR present three times in the PCR product generated from the major allele and four times in the product from the minor allele.
Results
Anatomical features
Under light microscopy the epithelium was of variable thickness and showed degenerate changes (Figure 1b), while Bowman's layer was replaced by a connective tissue pannus (Figure 1c). There were nodular, subepithelial, and anterior stromal deposits which stained positively with PAS, and showed an apple green birefringence with Congo red, typical of amyloid (Figure 1d). The stroma deep to the deposits was hypercellular.
Electron microscopy revealed that the epithelial cytoplasm was occupied at all levels by an abundant, fine, fibrillar, or filamentous material. Most epithelial cells showed features consistent with apoptotis, including condensation of nuclear chromatin and nuclear blebbing (Figure 2a). In some regions, basal cells were highly degenerate and showed advanced apoptotic changes (Figure 2b) with the formation of electron-dense bodies. Some basal cells possessed normal mitochondria (Figure 2c). Staining with cuprolinic blue showed the presence of proteoglycan (PG) within the cells and in the intercellular spaces between degenerate basal epithelial cells (Figure 2d). In places, rounded, vesicular structures were seen in relation to the basal lamina (Figure 2e). Bowman's layer was absent over the deposit.

Electron micrograph of GDLD cornea. (a) Apoptotic nucleus (N) in epithelium (E). (b) An advanced stage of apoptosis in a basal epithelial cell (E). (c) Normal mitochondria (M) in basal epithelial cell (E). (d) Proteoglycan deposits (PG) in intercellular spaces of epithelium (E). (e) Small vesicles (VS) in fibrous pannus below epithelium (E). (f) AM deposits in stroma. (g) Large proteoglycans filaments (PG) around amyloid deposits (AM). (h) Large proteoglycans (PG) near degenerate keratocytes.
A fibroblastic pannus was present beneath the epithelium, taking the place of Bowman's layer, and the subjacent superficial stroma was heavily infiltrated by 10–12 nm diameter fibrils typical of amyloid. Corneal fibroblasts within or near to the amyloid deposit were highly degenerate and showed prominent nuclear remnants and rough endoplasmic reticulum (RER). Usually, disorganised collagen fibrils were present in the region close to the amyloid deposits (Figure 2f). In the stroma outside the amyloid deposit zone, staining with cuprolinic blue showed large quantities of thick proteoglycan filaments (Figure 2g). The PG filaments were large in relation to degenerate keratocytes around the amyloid deposits (Figure 2h) and in places where collagen fibrils were sparse or degenerate. Throughout the stroma, the pannus and Bowman's layer, very fine microfibrils (1417 nm) were seen.
Expression of keratoepithelin (βig-h3)
Labelling was absent in immunoreaction controls of normal cornea where the primary antibody was omitted. In normal cornea, moderate labelling of the antibody was observed (Figure 3a). In GDLD cornea, little labelling for βig-H3 was found in the epithelium or in relation to Bowman's layer where this was intact. Moderate labelling by the antibody was seen on collagen fibrils (Figure 3b) and micro-fibrils in stroma (Figure 3c). Labelling on amyloid fibrils was sparse (Figure 3d).

Immunogold labelling of keratoepithelin and lactoferrin. (a) Immunoreaction control of normal cornea showing labeling in normal cornea. (b) Labelling of keratoepithelin in stroma (S) of GDLD cornea. (c) Labelling of keratoepithelin on microfibrils (MF) in stroma of GDLD cornea. (d) Scanty labelling on amyloid fibrils (AM) of GDLD cornea. (e) Labelling of lactoferrin in squamous cells of GLCD cornea. (f) Strong labelling of lactoferrin on amyloid fibrils (AM) of GLCD cornea.
Expression of lactoferrin
Lactoferrin immunolabelling was not observed in the normal cornea. In contrast, in the GDLD tissue, there was heavy labelling for lactoferrin both within and between the epithelial cells, with the strongest labelling in squamous and basal cells (Figure 3e) and less in the wing cells. Where Bowman's layer was present, labelling was sparse, but it was moderate within the pannus and strong in relation to the amyloid deposits (Figure 3f) and in the proximity of degenerate corneal fibroblasts. Only small amounts of labelling were seen in those keratocytes exhibiting a normal appearance. Sparse labelling was observed on the collagen fibrils or microfibrils.
Molecular genetics
The affected proband was homozygous for one known SNP (dbSNP rs14008; nucleotide A1263 C; nonsynonymous: amino acid E216 D) and one novel SNP (nucleotide A1133 C) within the M1S1 gene coding region, along with a novel G516 C, C517 G double-nucleotide polymorphism in a region corresponding to the M1S1 mRNA 5′ untranslated region (UTR). Since the novel A1133 C polymorphism introduced a D173 A amino-acid substitution in the coding region, it was considered a likely cause of GDLD in the affected proband. DNA was available from the mother and the maternal grandmother of the proband (Figure 4), who were found to be heterozygous (genotype AC) and homozygous (genotype AA) for the A1133 C SNP, respectively, consistent with recessive inheritance of a rare allelic variant (Figure 5). However, in a screen of 280 normal chromosomes, the C variant of the A1133 C SNP was found to occur at the relatively high frequency of 10.0% (Figure 6), in both Caucasian and Asian subjects. One of these control subjects was homozygous for the C variant.

Pedigree diagram. DNA for genotyping was available from subjects 1,3, and 7.

DNA sequencing traces showing the A1133 C polymorphism in the M1S1 gene of subjects: (a) affected proband, (b) mother of proband, and (c) grandmother of proband.

Agarose gel electrophoresis of Eae-I digestion products showing the A1133 C polymorphism of the M1S1 gene in subjects: Lane 1—affected proband, 2—mother of proband, 3—grandmother of proband, 4–14—control subjects, M—100 bp ladder.
Discussion
The subject of this study was a male Caucasian child with a bilateral, central, subepithelial avascular corneal disorder showing elevated gelatinous surface lesions typical of GDLD and exhibiting increased epithelial permeability to fluorescein. Deposits were shown to be due to the accumulation of amyloid. The features resembled those of gelatinous drop-like corneal dystrophy, an autosomal recessive disorder, due to mutations in the M1S1 gene. Vascularisation is a common feature of GDLD in reported series, but was not a feature of our case. However, there is significant phenotypic variation of this condition so that the absence of vascularisation does not exclude the diagnosis.6
GDLD shows both allelic and locus heterogeneity. Causative mutations in the M1S1 gene on chromosome 1p have been reported at positions M1R, C119 S, S170X, V194 E, Q118X, Q207X, 632delA and 870delC.9, 10, 12, 13, 15, 17, 23 Most subjects studied have been of Japanese or Indian origin, although Tunisian, American and European cases have also been reported.19, 24 Ren et al12 were able to exclude linkage to M1S1 in a single large Caucasian pedigree, confirming locus heterogeneity. Lactoferrin, a promising candidate gene could also be excluded by linkage analysis in this family.11 Mutations in the BIGH3 gene have been considered unlikely as a cause of GDLD,25 since of the wide variety of keratoepithelin mutations that have been documented, all exhibit autosomal dominant transmission.12
The affected proband was homozygous at three polymorphic sites in the M1S1 gene. One of these was in the 5′ UTR and thus considered unlikely to have functional consequences. The second was a known SNP at nucleotide position 1263 that has been reported previously in a Japanese cohort, (dbSNP rs14008; minor allele frequency 11%). The third was a novel A1133 C polymorphism that seemed a strong causative mutation candidate. However, further investigation revealed that this was also a relatively common SNP, with a minor allele frequency of approximately 10% in a sample of control subjects from the UK. Such a carrier frequency would give rise to a disease prevalence of ∼1% assuming full penetrance and random mating, and thus the A1133 C polymorphism could be excluded as the causative mutation in the affected proband. Since A1133 C SNP was not found in a cohort of 24 Japanese subjects in whom the rs14008 SNP was identified,26 it may be that the allele frequency of the A1133 C SNP is lower in Japan than the UK. Although theoretically possible, it is unlikely that combined homozygosity for the minor allelic variants of both the rs14008 and A1133 C SNPs is the cause of GDLD in the affected proband studied here. The close proximity of the two SNPs suggests that they would be in relatively strong linkage disequilibrium, in which case the frequency of homozygosity for both SNPs would be similar to that for either SNP alone. This would not be consistent with the rarity of GDLD.
The molecular mechanisms that give rise to the characteristic clinical features of GDLD are not yet known. One hypothesis is that the amyloid deposits arise by co-aggregation of the mutated M1S1 protein. There is evidence, too, that the amyloid protein colocalises with apolipoproteins -J and E, much as is found within Alzheimer plaques within the brain.27 However, its cellular source is unclear and we were unable to resolve this issue in the current study. Also, the reason for its accumulation in a manner that is different from that in other amyloid corneal dystrophies is not known.
A well-recognised feature of GDLD is an increase in epithelial permeability, associated with fluorescein staining of the ‘intact’ epithelium. This probably accounts for the intraepithelial accumulation of lactoferrin reported here and elsewhere.19 It has been assumed that the tears, which are rich in lactoferrin, are the source of the epithelial deposition.11 Quantock et al,28 demonstrated the ability of horseradish peroxidase, used as an in vitro tracer, to pass across the junctions between the most superficial epithelial cells, implying a functional defect in the epithelial tight junctions, which connect the surface cells of the corneal epithelium as zonulae occudentae. However, in our study, we found the accumulation to be intracellular, suggesting a defect in membrane permeability or an abnormal transport property of these cells. Although GDLD is not caused by a mutation in keratoepithelin, we were interested to know whether this adhesive protein might be incorporated into deposits in this patient. Stromal labelling of keratoepithelin is increased in other corneal disorders, such as bullous keratopathy, where it is assumed to be a reactive event. In our patient with GDLD, however, stromal labelling for keratoepithelin was not increased above normal, as assessed by immunoelectron microscopy. In the related corneal dystrophies granular, lattice, Avellino and honeycomb, the tissues are swamped with aggregates of the mutated keratoepithelin protein, which appears to form the basis for the dystrophic deposits. In other forms of corneal amyloid accumulation, the amyloid deposition is associated with an increase in stromal proteoglycan content.
The clinical and pathological features of this patient are in keeping with a diagnosis of GDLD. Ultrastructural examination suggested that the increased epithelial permeability may be the result of changes to the superficial epithelial cells than an absence of zonulae occludentae. An association between GDLD and developmental delay has not been reported previously, and while this may have been coincidental, a genetic basis seems likely. Our exclusion of the M1S1 gene as the disease locus in the affected proband is consistent with this hypothesis, and provides further evidence of locus heterogeneity in GDLD.
References
Buchi ER, Daicker B, Uffer S, Gudat F . Primary gelatinous drop-like corneal dystrophy in a white woman—a pathological, ultrastructural, and immunohistochemical study. Cornea 1994; 13: 190–194.
Lasram L, Rais C, Eleuch M, Quertani A . Gelatinous drop-like corneal-dystrophy. J Fr Ophtalmol 1994; 17: 24–28.
Kanai A, Kaufman HE . Electron-microscopic studies of primary band-shaped keratopathy and gelatinous, drop-like corneal-dystrophy in 2 brothers. Ann Ophthalmol 1982; 14: 535–539.
Fujiki K, Kanai A, Nakajima A . Frequency of gelatinous drop-like corneal dystrophy in Japanese population. Jpn J Med Genet 1986; 31: 210.
Fujiki K, Nakayasu K, Kanai A . Corneal dystrophies in Japan. J Hum Genet 2001; 46: 431–435.
Kinoshita S, Nishida K, Dota A, Inatomi T, Koizumi N, Elliott A et al. Epithelial barrier function and ultrastructure of gelatinous drop-like corneal dystrophy. Cornea 2000; 19: 551–555.
Ito M, Takahashi J, Sakimoto T, Sawa M . Histological study of gelatinous drop-like dystrophy following excimer laser phototherapeutic keratectomy. Nippon Ganga Ggakkai Zasshi 2000; 104: 44–50.
Tsubota K, Satake Y, Kaido M, Shinozaki N, Shimmura S, Bissen-Miyajima H et al. Treatment of severe ocular-surface disorders with corneal epithelial stem-cell transplantation. Arch Ophthalmol 2000; 118: 123.
Ha NT, Fujiki K, Hotta Y, Nakayasu K, Kanai A . Q118X mutation of M1S1 gene caused gelatinous drop-like corneal dystrophy: the P501T of BIGH3 gene found in a family with gelatinous drop-like corneal dystrophy. Am J Ophthalmol 2000; 130: 119–120.
Ha NT, Chau HM, Cung LX, Thanh TK, Fujiki K, Murakami A et al. A novel mutation of M1S1 gene found in a Vietnamese patient with gelatinous droplike corneal dystrophy. Am J Ophthalmol 2003; 135: 390–393.
Klintworth GK, Sommer JR, O’ Brian G, Han L, Ahmed MN, Qumsiyeh MB et al. Familial subepithelial corneal amyloidosis (gelatinous drop-like corneal dystrophy): exclusion of linkage to lactoferrin gene. Mol Vis 1998; 4: e31.
Ren ZX, Lin PY, Klintworth GK, Iwata F, Munier FL, Schorderet DF et al. Allelic and locus heterogeneity in autosomal recessive gelatinous drop-like corneal dystrophy. Hum Genet 2002; 110: 568–577.
Tasa G, Kals J, Muru K, Juronen E, Piirsoo A, Veromann S et al. A novel mutation in the M1S1 gene responsible for gelatinous droplike corneal dystrophy. Invest Ophthalmol Vis Sci 2001; 42: 2762–2764.
Tsujikawa M, Kurahashi H, Tanaka T, Okada M, Yamamoto S, Maeda N et al. Homozygosity mapping of a gene responsible for gelatinous drop-like corneal dystrophy to chromosome 1p. Am J Hum Genet 1998; 63: 1073–1077.
Tsujikawa M, Tsujikawa K, Maeda N, Watanabe H, Inoue Y, Mashima Y et al. M1S1 mutation in Japanese gelatinous drop-like corneal dystrophy. Invest Ophthalmol Vis Sci 2001; 42: 621.
Yamaguchi T, Fujiki K, Kanai A . Molecular genetic analysis of members of a single family with different types of corneal dystrophy (gelatinous drop-like corneal dystrophy and Avellino corneal dystrophy). Invest Ophthalmol Vis Sci 2001; 42: 2608.
Tsujikawa M, Kurahashi H, Tanaka T, Nishida K, Shimomura Y, Tano Y et al. Identification of the gene responsible for gelatinous drop-like corneal dystrophy. Nat Genet 1999; 21: 420–423.
Yoshida S, Kumano Y, Yoshida A, Numa S, Yabe N, Hisatomi T et al. Two brothers with gelatinous droplike dystrophy at different stages of the disease: role of mutational analysis. Am J Ophthalmol 2002; 133: 830–832.
Klintworth GK, Valnickova Z, Kielar RA, Baratz KH, Campbell RJ, Enghild JJ . Familial subepithelial corneal amyloidosis—a lactoferrin-related amyloidosis. Invest Ophthalmol Vis Sci 1997; 38: 2756–2763.
Munier FL, Korvatska E, Djemai A, LePaslier D, Zografos L, Pescia G et al. Kerato-epithelin mutations in four 5q31-linked corneal dystrophies. Nat Genet 1997; 15: 247–251.
Akhtar S, Bron AJ, Hawksworth NR, Bonshek RE, Meek KM . Ultrastructural morphology and expression of proteoglycans, βig-h3, tenascin-C, fibrillin-1, and fibronectin in bullous keratopathy. Br J Ophthalmol 2001; 85: 720–731.
Garcia-Closas M, Egan KM, Abruzzo J, Newcomb PA, Titus-Ernstoff L, Franklin T et al. Collection of genomic DNA from adults in epidemiological studies by buccal cytobrush and mouthwash. Cancer Epidemiol Biomark Prev 2001; 10: 687–696.
Nakamura T, Nishida K, Dota A, Adachi W, Yamamoto S, Maeda N et al. Gelatino-lattice corneal dystrophy: clinical features and mutational analysis. Am J Ophthalmol 2000; 129: 665–666.
Stock EL, Kielar RA . Primary familial amyloidosis of the cornea. Am J Ophthalmol 1976; 82: 266–271.
Klintworth GK . Advances in the molecular genetics of corneal dystrophies. Am J Ophthalmol 1999; 128: 747–754.
Haga H, Yamada R, Ohnishi Y, Nakamura Y, Tanaka T . Gene-based SNP discovery as part of the Japanese Millennium Genome Project: identification of 190,562 genetic variations in the human genome. Single-nucleotide polymorphism. J Hum Genet 2002; 47: 605–610.
Nishida I, Quantock AJ, Dota A, Choi-Miura NH, Kinoshita S . Apolipoproteins J and E co-localise with amyloid in gelatinous drop-like and lattice type I corneal dystrophies. Br J Ophthalmol 1999; 83: 1178–1182.
Quantock AJ, Nishida K, Kinoshita S . Histopathology of recurrent gelatinous drop-like corneal dystrophy. Cornea 1998; 17: 215–221.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Akhtar, S., Bron, A., Qin, X. et al. Gelatinous drop-like corneal dystrophy in a child with developmental delay: clinicopathological features and exclusion of the M1S1 gene. Eye 19, 198–204 (2005). https://doi.org/10.1038/sj.eye.6701453
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.eye.6701453
