
Abstract
A family was previously reported as suffering from severe granular dystrophy. The phenotypic picture suggested a mix of homozygous and heterozygous family members. Genetic analysis confirms the homozygousity in the patients most severely affected, but shows the disease state to be one of Avellino corneal dystrophy. The previous case reports are extended immunohistological staining using polyclonal antibodies raised against keratofepithelin. This genotype/phenotype correlation study is consistent with incomplete dominance.
Similar content being viewed by others
Introduction
A previously reported1 Asian pedigree of severe granular dystrophy, where three members were postulated as being homozygotes, was re-examined. At the time of the first report, the pedigree was too small for linkage analysis. It has been shown2 that distinct BIGH 3 mutations cause six 5q31-linked corneal dystropies, that is, granular type I (Groenouw type I), granular type II (Avellino), granular type III (Reis–Bücklers), honeycomb (Thiel–Behnke), lattice type I (Biber–Haab), and lattice type IIIa. These mutations are R555W, R124H, R124L, R555Q, R124C, and P501T, respectively. Our consanguineous pedigree was molecularly characterised and clinically reassessed in order to further delineate the spectrum of phenotypes associated with the segregation of a specific BIGH 3 mutant. In addition, a corneal button from a homozygously affected family member was used for immunostaining with anti-KE antibodies.
Methods
Family members were approached to take part in the current study. This involved an examination of the corneas and the taking of a blood sample for genetic analysis. Where appropriate, case records were examined. In addition, previous penetration keratoplasty pathology specimens were re-examined.
For genetic analysis, 5 ml of blood in an EDTA tube was taken after informed consent. After DNA extraction, exon 4 was PCR-amplified and bidirectionally sequenced on an ABI 310′ sequencer. One previous pathology specimen from case 1 was located. Sections were taken and immunostaining using KE-15 and KE-2 was performed as previously described.3
Case report updates
Case 1
This patient presented at the age of 14 years in 1970. At that time, he had a severe corneal dystrophy and nystagmus. Best-corrected acuity was 6/18 on the right and 6/36 on the left eye with contact lenses. Lammellar corneal grafts were performed in 1975 and thereafter, best-corrected vision was 6/18 right and 6/24 left. After 2½ years, there was recurrence in both grafts, between 1980 and 1990, these were two penetrating grafts in each eye. The maximum visual acuity was 6/12 on the right and 6/24 on the left eye. Since 1992, phototherapeutic keratectomy (PTK) was used 5 times in the right and 4 times in the left. This has maintained visual acuities at 6/24 best corrected in either eye. This patient is awaiting further PTK to the right cornea.
Case 2
This patient presented at the age of 8 years in 1970. Visual acuities were 6/6 on the right and 6/18 on the left eye. The main problems were ones of recurrent erosions and photophobia. These were not helped by an episode of bilateral dendritic ulcers in 1978 with recurrence of the dendrites thereafter. Between 1980 and 1990, the right eye has been grafted twice and the left eye once. Best-corrected acuity was 6/6 right and 6/9 left. Since 1992, PTK has been used twice on the right and four times on the left eye. This has maintained visual acuities of 6/9 in either eye. This patient is a waiting further treatment to his right cornea.
With both these cases, recurrence of the dystrophy was within 4–6 months. By staggering treatments between the eyes, these patients are having PTK at about yearly intervals.
Case 3
This patient presented in 1973, aged 10 years with superficial corneal dystrophy. The referral letter stated that the father suffered from lattice corneal dystrophy. Visual acuity was 6/24 in each eye. Between 1978 and 1990, the left eye has had two penetrating grafts and one lamellar graft; the right eye has had two penetrating grafts. The grafting has been complicated by glaucoma and refractive problems. Best-corrected vision is 6/18 with contact lenses. PTK has only been indicated for the right cornea as the left has deep vascularisation and scarring of the grafts below the level of the corneal deposits. A right PTK has been performed with the best-corrected visual acuity of 6/36. Attempts at scleral contact lens wear have not improved visual acuity. The results have been disappointing due to the post-corneal grafting irregular refractive changes.
Case 4
This patient aged 49 years was referred in 1982 by her optician, with granular dystrophy. Visual acuity was 6/6 in either eye. Over the next 4 years, vision deteriorated to 6/60 and the patient was offered grafting. Since 1992, there have been symptoms of recurrent erosions with a best visual acuity of 6/24. Symptoms only require topical medical treatment at present.
Results
In total, 18 patients of the pedigree were examined and blood samples were obtained from 17 of the available family members (Figure 1). Upon sequencing of exon 4, an R124H mutation was identified to cosegregate with all affected family members. All patients were heterozygous for this mutation, except the severely affected individuals, who were homozygous carriers. Illustrative corneal photographs are shown in Figures 2 and 3. Immunochemistry and histology are shown in Figure 4.

Pedigree with genotype and phenotype.

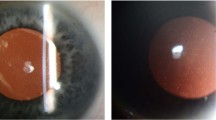
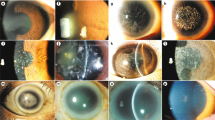
Heterozygote patient showing progression of corneal changes over 3 years (left eye) (a). Arrowed linear deposit in latest image (b).

Case 2 homozygote (IIC left eye) showing changes prephototherapeutic keratectomy (a) and after four treatments (b).

Tissue sections of the corneas from case 1 (IIA) deposit arrowed, asterisked staining in epithelium: (a) haematoxylin and eosin staining; (b) immunostaining with ati Amyloid P antibodies; (c) immunstaining using antibodies against KE (KE-15); (d) immunstaining using antibodies against KE (KE-2).
The clinical appearances of the homozygote patients are difficult to interpret due to the repeated treatments. The recurrence at the suture tracts is usual in granular dystrophy. There are no signs of lattice. There is corneal haze, but this may be secondary to their treatment. The eldest heterozygote patient shows a superficial dystrophy with rings of granular material. There is one linear patch on the right cornea that could be construed as a lattice deposit.
One histology specimen was examined from case 1; this was following the first perforating keratoplasty performed in the right eye following previous right lamellar graft, that was homozygously affected. The subepithelial deposits localise at the level of the Bowman's layer that is disrupted (Figure 4a). Staining for amyloid P is negative (Figure 4b). There was a strong immunoreactivity of the subepithelial deposits with KE-15 and KE-2 (Figure 4c and d, respectively). Keratoepithelin staining was also noticed within the interstitial space at the level of the basal and intermediate cell layer of the epithelium.
Discussion
In retrospect, the older heterozygous members of this pedigree show the anterior stromal ring opacities associated with the granular type II (Avellino) phenotype. These opacities are large and few in number in Avellino dystrophy compared to the small, numerous deposits in granular dystrophy. In addition, in Avellino dystrophy, there may be other deposits with a crystal-like or snowflake appearance. The homozygous members prior to their surgeries were described as having a superficial severe granular dystrophy. Early histology reports confirmed this diagnosis.
The term Avellino corneal dystrophy was coined in a report of two related Italian–American families whose origin was in the town of Avellino in Campania, Italy.4 Avellino corneal dystrophy has previously been reported as granular-lattice dystrophy.5 It has since been reported in pedigrees with various ethnic origins, including Japanese patients.6, 7
In Avellino corneal dystrophy, there are features of both granular and lattice dystrophy. The lesions in Avellino are typically large, grey ringed or snowflake in appearance in the anterior stroma with lattice-like lesions developing later and deeper. The number of lesions are typically less than that seen in granular dystrophy, where they may be too numerous to count. The dystrophic process progresses with age; typically younger patients have a more granular pattern that tend to stabilise early with a gradual production of lattice-like patterns in later life.8 The stroma between lesions can be hazy, although this has not been a feature of this reported pedigree.1, 5 Vision is relatively unaffected early in life. In this pedigree, the eldest heterozygote has minimal acuity changes despite the extensive corneal changes. The most common problem is one of recurrent erosions due to the superficial nature of the granular deposits. The homozygous members of this pedigree have attended the ophthalmic casualty frequently for treatment of recurrent erosions. This is less of a problem for patients with granular dystrophy. There is phenotypic variability within families with Avellino dystrophy, with some individuals developing granular changes alone by 6 or 32 years of age.5, 8 This variability is also seen in histological specimens with some cases of Avellino having no amyloid detected in the cornea.9
Homozygosity for the R124H and R555W mutations has been reported previously.9, 10, 11 In one report of homozygosity in Avellino dystrophy, the heterozygotes were diagnosed clinically as granular dystrophy.10 Histologic studies in another report could not find amyloid deposits in the corneal buttons despite a R124H mutation suggesting Avellino corneal dystrophy.9 In one report of homozygosity in granular dystrophy, the genetic analysis confirmed an R555W mutation;11 the proband who presented at 6 years of age, required five keratectomies over a 19-year follow-up period.
Corneal grafting has been the usual treatment for visual improvement in severe corneal dystrophies. In heterozygous corneal dystrophies, recurrence in a graft can take 4–33 months to develop.12 Clinically significant recurrence (which caused a decrease in vision) can take, on an average, 59 months to develop.13 In these homozygote cases, recurrence has taken 12–24 months. There is a link between the genetic molecular defect and the clinical course of corneal dystrophies, so that genetic characterisation could help to predict future clinical progress in ambiguous cases.14 Phototherapeutic keratectomy has been used for superficial corneal lesions and scars.15 This is a useful and, especially with these patients reported, a repeatable treatment that maintains visual acuity.
Gene analysis has confirmed the previously reported clinical impression of homozygosity in this pedigree.1 It was also instrumental in better refining the diagnosis as being granular dystrophy type II (Avellino). Immunohistology staining consistently identified mutant KE as a major component of the corneal deposits. The classification of these two anterior stromal dystrophies should be ultimately based on gene analysis. However, one can see that there is a phenotypic difference between these two conditions on clinical and histological grounds. In severe cases of anterior corneal dystrophies, other family members should be examined and gene analysis performed to distinguish between the homozygous states of these two conditions. The term ‘juvenile superficial granular dystrophy’ and its variants should not be used unless gene analysis shows mutations other than those seen with granular or Avellino dystrophy.
This report has also discussed the management of these severe cases. PTK is less invasive than penetrating keratoplasty with lower morbidity and is the initial treatment of choice. Gene analysis or immunochemistry may be useful in cases where the clinical features of the corneal dystrophy may not be conclusive.
References
Diaper CJM . Severe granular dystrophy: a pedigree with presumed homozygotes. Eye 1994; 8: 448–452.
Munier FL, Korvatska E, Djemai A, Le Paslier D, Zografos L, Pescia G et al. Kerato-epithelin mutations in four 5q31-linked corneal dystropies. Nature Gene 1997; 15: 247–251.
Korvatska E, Munier FL, Chaubert P, Wang MX, Uffer S, Zografos L et al. Kerato-epithelin, a new amyloidogenic protein in corneal dystrophies. Inv Ophthalmol Vis Sci 1999; 40: 2213–2219.
Holland EJ, Daya SM, Stone EM, Folberg R, Dobler AA, Cameron JD et al. Avellino corneal dystrophy. Ophthalmology 1992; 99: 1564–1568.
Folberg R, Alfonso E, Croxatto JO, Driezen NG, Panjwani N, Laibson PR et al. Clinically atypical granular dystropgy with pathologic features of lattice like amyloid deposits. a study of 3 families. Ophthalmology 1988; 95: 46–51.
Kennedy SM, McNarmara M, Hillery M, Hurley C, Collum LMT, Giles S . Combined granular lattice dystrophy (Avellino corneal dystrophy). Br J Ophthalmol 1996; 80: 489–490.
Dolmetsch AM, Stockl FA, Folberg R, Gensini I, Burnier Jr MN . Combined granular lattice dystrophy (Avellino) in a patient with no known Italian ancestry. Can J Ophthalmol 1996; 31: 29–31.
Rosenwasser GOD, Sucheski BM, Rosa N, Pastena B, Sebastiani A, Sassani JW et al. Phenotypic variation in combined granular lattice (avellino) corneal dystrophy. Arch Ophthalmol 1993; 111: 1546–1551.
Okada M, Yamamoto S, Inoue Y, Watanabe H, Maeda N, Shimomura Y et al. Severe corneal dystrophy phenotype caused by homozygous R124H keratoepithelin mutations. Invest Ophthalmol Vis Sci 1998; 39: 1947–1953.
Fujiki K, Hotta Y, Nakayasu K, Kanai A . Homozygotic patient with beta IG-h3 gene mutation in granular dystrophy. Cornea 1998; 17: 288–292.
Okada M, Yamamoto S, Watanabe H, Shimomura Y, Tano Y . Granular corneal dystrophy with homozygous mutations in the kerato-epithelin gene. Am J Ophthalmol 2000; 129: 411–412.
Lyons CL, McCartney AC, Kirkness CM, Ficker LA, Steele ACMcG, Rice NSC . Granular corneal dystrophy—visual results and pattern of recurrence after lamellar or penetrating keratoplasty. Ophthalmology 1994; 101: 1812–1817.
Marcon AS, Cohen EJ, Rapuano CJ, Laibson PR . Recurrence of corneal stromal dystrophies after penetrating keratoplasty. Cornea 2003; 22: 19–21.
Ellies P, Renard G, Valleix S, Boelle PY, Dighiero P . Clinical outcome of eight BIGH3-linked corneal dystrophies. Ophthalmology 2002; 109: 793–797.
Maclean H, Robinson LP, Wechsler AW, Goh A . Excimer phototherapeutic keratectomy for recurrent granular dystrophy. Aus N Z J Ophthalmol 1996; 24: 127–130.
Acknowledgements
We acknowledge the enthusiastic help of all the family members with this study. FL Munier acknowledges the support from the Swiss National Science Foundation (Grant # 32-065250.01). No propriety interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Diaper, C., Schorderet, D., Chaubert, P. et al. Clinical and immunopathological corneal phenotype in homozygotes for the BIGH3 R124H mutation. Eye 19, 92–96 (2005). https://doi.org/10.1038/sj.eye.6701398
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.eye.6701398
Keywords
This article is cited by
-
Identification of two novel mutations in the cornea-specific TGFBI gene causing unique phenotypes in patients with corneal dystrophies
International Ophthalmology (2016)
