
Abstract
Targeted therapy has radically altered the way metastatic renal cancer is treated. Six drugs are now licensed in this setting, with several other agents under evaluation. Sunitinib is currently the most widely used in the first line setting with impressive efficacy and an established toxicity profile. However, as further randomised studies report and as newer drugs become available this may change. In this review, we address our current understanding of targeted therapy in renal cancer. We also discuss areas in which our knowledge is incomplete, including the identification of correlative biomarkers and mechanisms of drug resistance. Finally, we will describe the major areas of clinical research that will report over the next few years.
Similar content being viewed by others
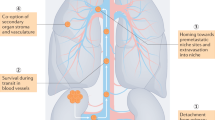

Main
The treatment of metastatic clear cell renal carcinoma (RCC) has dramatically changed over the last 5 years. This has been driven by two groups of targeted agents; namely vascular endothelial growth factor (VEGF)-targeted therapies and mammalian target of rapamycin (mTOR) inhibitors. Both, first and second line treatment is of proven benefit and these agents have replaced immune therapies that were previously standard of care for metastatic RCC (Motzer and Bukowski, 2006; Motzer et al, 2009).
Sunitinib is a multitargeted tyrosine kinase inhibitor (TKI) that predominantly targets VEGF (Figure 1). It also has ‘off target’ effects, involving other tyrosine kinases that may account for some of its activity and toxicity (Motzer and Bukowski, 2006). The pivotal trial of sunitinib was published in 2006; and subsequently it has established itself as standard first line therapy (Motzer et al, 2009). The exact mechanism of its activity in RCC remains unknown, and it is not yet possible to identify specific cohorts of patients who benefit from therapy. Additionally, sunitinib and the other drugs are only effective in controlling the disease for a limited period before progression occurs. Therefore, an important area of research is investigating mechanisms of resistance.

Diagrammatic representation of the mechanism of action of targeted therapy in clear cell renal cancer. This figure shows how HIF-1 is unregulated within the clear cell renal cancer tumour cell by two mechanisms. This upregulation in turn stimulates receptors on endothelial cells (VEGF and PDGF). Dotted lines denote the drug targets. Off-target effects on the stroma and tumour cells are not represented.
Sunitinib is one of a number of VEGF-targeted agents with activity in this setting. Four drugs including sunitinib have been approved by the FDA for the treatment of metastatic RCC. The other three agents are sorafenib, pazopanib and bevacizumab (Escudier et al, 2007a, 2007b; Rini et al, 2008a, 2008b; Sternberg et al, 2009). Newer agents under investigation in ongoing randomised trials include tivozinib, dovitinib and axtitinib. It is hoped that these agents will show improved activity, decreased toxicity or set a benchmark as second or third line therapy. However, none of these trials are aimed at specific subsets of patients with metastatic clear cell RCC.
The other major group of targeted agents in renal cancer is the mTOR inhibitors. These include temsirolimus and everolimus that are both widely used and have proven activity (Hudes et al, 2007; Motzer et al, 2008a, 2008b). Once again there has been a failure to identify specific groups of patients that benefit from these agents, and although these agents offer initial benefit, resistance occurs.
This review addresses our current understanding of the role of targeted therapy in renal cancer. It will also highlight areas in which our understanding is incomplete and finally will describe the major areas of clinical research, which will report over the next few years.
Histology and prognosis in metastatic renal cancer
Renal cell carcinoma consists of a number of histological subtypes, the most common of which is clear cell RCC. It is characterised by a mutation to the Von Hippel Lindau protein that is linked to both regulation of HIF and VEGF (Na et al, 2003) (Figure 1). Papillary RCC accounts for the bulk of the remaining ‘non-clear cell’ tumours. The majority of trials focus on the clear cell population in which HIF/VEGF appears to be the dominant tumour biology (Rini et al, 2008a, 2008b).
The most commonly used method for predicting outcome of patients with metastatic renal cancer is the Memorial Sloan-Kettering Prognostic score. It stratifies patients into three groups (good, intermediate and poor) depending on a number of factors, including lactate dehydrogenase, performance status, serum calcium, haemoglobin and time since diagnosis to treatment (Motzer and Bukowski, 2006). Although designed before the introduction of targeted therapy it has been validated in the TKI era (Motzer et al, 2008; Heng et al, 2009).
Sunitinib in metastatic renal cancer
Sunitinib is superior to interferon in the first line treatment of clear cell RCC. In the pivotal study the progression-free survival (PFS) was 11 months for sunitinib vs 5 months for interferon (hazard ratio (HR): 0.53; (95% CI: 0.45–0.64); P<0.001) (Motzer et al, 2007, 2009). Most of the study patients were MSKCC good and intermediate risk, had a good performance status and a nephrectomy before treatment. The majority of patients on the interferon arm subsequently received sunitinib (33%) or other VEGF-signalling inhibitors (32%) confounding the overall survival (OS) data (26.4 months for sunitinib vs 21.8 months for interferon (HR: 0.821; 95% CI: 0.673–1.001; P=0.051). Subsequent analysis of those patients who did not cross over to sunitinib showed an OS of 14 months for interferon and 28 months for sunitinib (Flanigan et al, 2001; Motzer et al, 2009). The benefits of sunitinib were present in all three MSKCC prognostic groups, although the numbers in the poor-risk category were small (<10% of the study population) resulting in statistically insignificant data. Subsequent data from a large expanded access series reproduced the PFS results (10.9 months; 95% CI: 10.3–11.2 months), with an OS of 18.4 months (95%CI: 17.4–19.2 months) underlining the practise changing nature of these results (Gore et al, 2009).
There is a lack of randomised data for sunitinib in the non-clear cell population. The most comprehensive data comes from the expanded access cohort in which the PFS and OS were 7.8 (95% CI: 6.3–8.3) months and 13.4 (95% CI: 10.7–14.9) months, respectively (Gore et al, 2009). A further retrospective analysis supported the use of sunitinib in this setting with a PFS of 11.9 months (Choueiri et al, 2008).
The most common toxicities encountered with sunitinib included (grade 3 or more) hypertension (12%), fatigue (11%), diarrhoea (9%) and hand–foot syndrome (9%). In the landmark phase III study 50% of the patients required a dose reduction of sunitinib due to these events (Motzer et al, 2008a, 2008b). A recent meta-analysis identified a relationship between steady-state area under the curve of sunitinib and time to tumour progression, toxicity and OS (Houk et al, 2009) suggesting increased exposure to sunitinib appears to be associated with improved clinical outcomes, as well as increased risk of adverse effects. Therefore, attempts should initially be made to address toxicity before dose reductions are made. Sunitinib is currently given at 50 mg for 4 consecutive weeks followed by a 2-week treatment-free interval. Phase II data investigating continuous dosing at 37.5 mg showed similar toxicity profile to the 50 mg intermittent dose (Escudier et al, 2009a, 2009b, 2009c). A randomized phase II study comparing the standard 50 mg regimen with continuous 37.5 mg dosing is exploring this regimen formally and will report in 2010.
Predicting clinical benefit with targeted therapy
A number of prognostic biomarkers have been identified that correlate with a poor outcome with sunitinib. These include the MSKCC prognostic criteria, raised platelet/neutrophil levels at diagnosis (Heng et al, 2009; Motzer et al, 2008a, 2008b). However, it has not been possible to identify biomarkers or imaging modalities that predict clinical benefit with specific agents. Sunitinib causes dynamic changes to cytokine expression and growth factors; however, it remains unclear whether these predict clinical benefit (Rini et al, 2008a, 2008b). Further work in this area is required.
Although widely used, response rates using CT (RECIST criteria) do not correlate with outcome (Kontovinis et al, 2009). Other methods of correlating CT changes with outcome, such as the Choi criteria, which measures tumour attenuation (Hounsfield units), as well as changes in two-dimensional size, are under investigation and hold promise (van der Veldt et al, 2010). Other imagining modalities, such as dynamic contrast enhanced MRI and FDG-PET, are also being investigated (Hahn et al, 2008).
Perhaps the most promising current biomarker is the presence of treatment-associated hypertension, which in retrospective analysis, appears to correlate with outcome (Rixe et al, 2008). This finding is under investigation in prospective studies with axitinib. It raises a number of issues, for example, is this finding a pharmacokinetic or pharmacodynamic affect and if the hypertension is associated with clinical benefit how aggressively should it be managed?
The optimal way of identifying predictive rather than prognostic biomarkers in this setting is with prospective studies comparing VEGF-targeted agents with other classes of drug, such as the mTOR inhibitors. Using this approach it is possible to determine whether subgroups benefit from the specific agents rather than just identifying prognostic markers. Clinical trials such as RECORD 3 that compares sunitinib and everolimus are the closest we have to this design, although the identification of biomarkers in not the primary endpoint and the translational component is, therefore, not comprehensive.
Other VEGF-targeted agents
(Table 1) Sorafenib and bevacizumab were both developed at approximately the same time as sunitinib in RCC. Sorafenib is associates with a PFS benefit as second line therapy after interferon failure (Escudier et al, 2007a, 2007b). However, a first line sorafenib vs interferon randomised phase II study showed no benefit for sorafenib, making it less attractive in this setting (Escudier et al, 2009a, 2009b, 2009c).
Two randomised phase III studies investigated bevacizumab in combination with interferon vs interferon alone as first line treatment for metastatic RCC (Escudier et al, 2007a, 2007b; Rini et al, 2008a, 2008b). These studies focused on the clear cell population and consisted of predominantly good and intermediate risk patients. Both studies showed a benefit for the combination in terms of PFS (5.1 vs 8.5; 5.4 vs 10.2), with the OS for in the bevacizumab arm of 18.3 and 22.9 months ref.
More recently Pazopanib has received FDA and EMA approval. In the pivotal randomised phase III study the PFS for the pazopanib was 11.1 months compared with 2.8 months for placebo for previously untreated patients (HR: 0.40; 95% CI: 0.27, 0.60; P<0.001) (Sternberg et al, 2009). The toxicity profile for pazopanib may be different to the other VEGF TKIs with the most common adverse event being diarrhoea (52%; 4% Grade 3 or 4), hypertension (40%; 4% Grade 3 or 4), hair colour changes (38%) and nausea (26%; <1% Grade 3 or 4). The incidence of fatigue and ‘hand and foot syndrome’ appears to be relatively low (grade 3 or more in 5 and 2%, respectively). However, abnormalities in liver function tests (>3 × normal) occurred in 20% of patients (Sternberg et al, 2009; Hutson et al, 2010). These factors may help select the optimal agent for individuals. A pivotal study comparing pazopanib and sunitinib in the first line setting has closed recruitment and will report in 2011.
Although direct comparison of the above agents is not currently possible because of the lack of direct randomized studies in the first line setting, indirect comparison is possible but flawed in nature (Table 1). Nevertheless sunitinib has set a benchmark for both overall and PFS here, although the PFS for pazopanib is also 11 month and is, therefore, promising. In view of the fact that sorafenib did not appear superior to interferon it is more difficult to recommend this agent above those with proven superiority to interferon, such as bevacizumab (with interferon) and sunitinib (Escudier et al, 2009a, 2009b, 2009c).
mTOR inhibitors
Everolimus and temsirolimus are both mTOR inhibitors (TORC1) that have been investigated and are widely used in metastatic RCC (Hudes et al, 2007; Motzer et al, 2009) (Figure 1). Temsirolimus was investigated in untreated poor risk disease. Results of this study showed prolonged OS for temsirolimus compared with interferon (HR for death: 0.73; 95% CI: 0.58–0.92; P=0.01). However, the combination of temsirolimus and interferon together did not show significant benefit over temsirolimus alone. It is speculated that this may be because of the dose intensity achieved in this arm of the study. Subset analysis showed significant benefit for non-clear cell population that is of particular interest and warrants further investigation. The drug was relatively well tolerated with fewer serious adverse events in the temsirolimus group than in the interferon group (P=0.02).
Everolimus is the only agent with positive randomised data after TKI failure in RCC (Motzer et al, 2009). RECORD 1 compared the effects of everolimus and placebo in patients who had previously received at least one line of targeted therapy. The PFS strongly favoured everolimus (HR: 0.30; 95% CI: 0.22–0.40; P<0.0001). Perhaps, the most significant toxicity seen was pneumonitis (any grade 8%), which requires particular attention with using this agent.
VEGF inhibitors as second line therapy in RCC
Although phase III data support the use of sorafenib as second line therapy, this study conducted in the era of immune therapy and (Escudier et al, 2007a, 2007b), there is no randomised data to support further VEGF TKI therapy after failure of initial targeted therapy. However, phase II data suggest that there may be non-cross resistance between these agents (Rini et al, 2009; Sablin et al, 2009). Therefore, the use of sequential VEGF-targeted therapy may be beneficial. A number of randomised phase II and III studies, which investigate a switch from one TKI to another, are ongoing. Pazopanib, sorafenib, axitinib, cediranib and sunitinib are all being investigated in this setting. Data with axitinib is likely to be the first of these studies to report. Axitinib is a potent VEGF-specific target TKI with few ‘off target’ effects. Results in the post interferon setting are impressive (PFS of 15.7 months (95% CI: 8.4–23.4)) and a phase II study that investigated axitinib post sorafenib are particularly striking (PFS and OS were 7.4 months (95% CI: 6.7–11.0) and 13.6 months (95% CI: 8.4–18.8), respectively) (Rixe et al, 2007; Rini et al, 2009). A randomised phase III second line study, comparing sorafenib and axitinib post VEGF-targeted therapy is now closed and results are awaited. The occurrence of hypertension with axitinib may predict clinical benefit, which is being investigated formally in clinical trials (Rixe et al, 2008).
Other agents under investigation
Tivozinib is a potent VEGF TKI with impressive clinical data. In a randomised discontinuation study 88% of patients obtained a clinical benefit from the drug (Bhargava et al, 2009). A randomised phase III study comparing tivozinib and sorafenib in patients not previously exposed to targeted therapy will close in December 2010. Dovitinib is another promising TKI that targets FGF-2, as well as VEGF. It is being investigated in the third line setting in mTOR refractory disease.
Combination therapy
A logical step in improving outcomes is to investigate these drugs in combination. However, combining sunitinib with other forms of targeted therapy has been difficult because of excessive toxicity (Feldman et al, 2009). This may not be the case with bevacizumab, which has been shown to be tolerable in combination with other agents such as everolimus. In a phase II study of bevacizumab and everolimus the median PFS for this combination was 9.1 months with response rates of 30% (Hainsworth et al, 2010). A recent randomised phase II study comparing sunitinib v.s. bevacizumab and interferon v.s bevacizumab and temsirolimus demonstrated significant toxicity for the latter combination without any efficacy advantage (Escudier et al, 2010). Perhaps, the most early awaited combination data is RECORD II that compares bevacizumab and everolimus vs bevacizumab and interferon. The potential disadvantages of combination therapy include additional toxicity, cost and the theoretical risk of multidrug resistance. Early results in this setting have been disappointing.
Mechanisms of resistance to therapy
The mutation to the VHL gene and subsequent activation of HIF and VEGF are hall marks of clear cell renal cancer. The VEGF-targeted therapy is thought to work by blocking the angiogenic effects of this overactive pathway (Gossage and Eisen, 2010). Only a small minority of renal tumours initially progress through VEGF-targeted therapy, and the majority of patients initially obtain clinical benefit with these drugs (Motzer et al, 2009). However, resistance occurs, which is usually referred to as acquired resistance (Rini and Atkins, 2009). The onset of this acquired resistance is variable both in terms of time and clinical pattern, and our understanding of it at the molecular level is in its infancy. This is largely due to the lack of sequential tissue for comparative analysis.
Unlike in other malignancies, the acquisition of specific mutation to the target of the drug is not thought to be responsible for resistance in RCC (Valent, 2008; Rini et al, 2009). This is because in RCC the main target for sunitinib is the VEGF receptors on the vascular endothelium that are genetically stable.
There are currently two main theories to account for acquired resistance. First, it is speculated that ‘angiogenic escape’ occurs, in which with time the initial targeted therapy becomes ineffective at blocking the VEGF axis. This hypothesis is supported by the observation that switching from one VEGF-targeted therapy to another potentially more potent agent results in further responses (Rini et al, 2009). It is speculated that this may be due to upregulation of associated growth factors, such as HIF1-α (Rini et al, 2009). The second area of investigation is based around the emerging preclinical data that support the role of alternative pathways and growth factors in propagating tumour growth in acquired resistance. Specifically, FGF-2, tie2/Ang2, il-8 and the Src family have been implicated in this process (Oliner et al, 2004; Cenni et al, 2005; Bergers and Hanahan 2008). Clinical trials have been designed to target all these protein with dovotinib, AMG386 and saracatinib. Resistance also occurs universally with the mTOR inhibitors. The current drugs in mRCC only target TORC1 and it appears that inhibition of this in isolation leads to compensatory upregulation of PI3 kinase (O’Reilly et al, 2006). The development of combined TORC1 and 2 inhibitors could potentially overcome this and will be investigated in mRCC (Sarbassov et al, 2006).
Clinical trials in the future
The two main goals over the next 5–10 years in mRCC are to firmly establish the most effective agents in each class and identify predictive biomarkers associated with clinical benefit to specific agents. To achieve these goals two different trial designs are required. Randomised phase III studies comparing newer agents with benchmark controls (currently sunitinib for the first line and everolimus for second line) are required to redefine standard therapy. The COMPARZ study, a non-inferiority study, which compares pazopanib and sunitinib, has just closed and is due to report in 2011/12. Results are eagerly awaited by further studies with tivozinib and axitinib vs a benchmark are required before there is clarity about the optimal first line VEGF TKI. The assumption that these agents are more active in the first-line setting without the appropriate studies is flawed, despite their impressive efficacies in other settings. Sorafenib should not be considered an adequate benchmark control in the first line setting (Escudier et al, 2009a, 2009b, 2009c).
There is also a need for smaller biomarker studies, which take sequential tissue, plasma and functional imaging during therapy. These studies are aimed at gaining a better understanding of how each drug works. The true goal for these studies is to identify predictive biomarkers for specific agents, which requires randomisation against other agents.
Conclusions
Targeted therapy has changed the way metastatic renal cancer is treated and the OS for these patients is now greater than 2 years in prospective studies. Sequential therapy with VEGF-targeted therapy and mTOR inhibitors are currently the standard of care. Data on specific combinations are eagerly awaited. Correlative markers associated with clinical benefit remain elusive and are urgently required to develop treatment for this disease. This will help us to move away from the current ‘one size fits all’ approach and help develop truly individualised targeted therapy.
Change history
29 March 2012
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Bergers G, Hanahan D (2008) Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer 8: 592–603
Bhargava P, Esteves B, Nosov DA, Lipatov ON, Lyulko AA, Anischenko AA, Chacko RT, Lee P, Al-Adhami M, Ryan J (2009) Updated activity and safety results of a phase II randomized discontinuation trial (RDT) of AV-951, a potent and selective VEGFR1, 2, and 3 kinase inhibitor, in patients with renal cell carcinoma (RCC). ASCO Ann Meet Proc 27 (Suppl): 15S (abstract 5032)
Cenni E, Perut F, Zuntini M, Granchi D, Amato I, Avnet S, Brandi ML, Giunti A, Baldini N (2005) Inhibition of angiogenic activity of renal carcinoma by an antisense oligonucleotide targeting fibroblast growth factor-2. Anticancer Res 25 (2A): 1109–1113
Choueiri TK, Plantade A, Elson P, Negrier S, Ravaud A, Oudard S, Zhou M, Rini BI, Bukowski RM, Escudier B (2008) Efficacy of sunitinib and sorafenib in metastatic papillary and chromophobe renal cell carcinoma. J Clin Oncol 26: 127–131
Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Siebels M, Negrier S, Chevreau C, Solska E, Desai AA, Rolland F, Demkow T, Hutson TE, Gore M, Freeman S, Schwartz B, Shan M, Simantov R, Bukowski RM, TARGET Study Group (2007a) Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med 356: 125–134
Escudier B, Negrier S, Gravis G, Chevreau C, Delva R, Bay J, Geoffrois L, Legouffe E (2010) Can the combination of temsirolimus and bevacizumab improve the treatment of metastatic renal cell carcinoma (mRCC)? Results of the randomized TORAVA phase II trial. J Clin Oncol 28 (Suppl): 7s (abstract 4516)
Escudier B, Pluzanska A, Koralewski P, Ravaud A, Bracarda S, Szczylik C, Chevreau C, Filipek M, Melichar B, Bajetta E, Gorbunova V, Bay JO, Bodrogi I, Jagiello-Gruszfeld A, Moore N, AVOREN Trial investigators (2007b) Bevacizumab plus interferon alfa-2a for treatment of metastatic renal cell carcinoma: a randomised, double-blind phase III trial. Lancet 370: 2103–2111
Escudier B, Roigas J, Gillessen S, Harmenberg U, Srinivas S, Mulder SF, Fountzilas G, Peschel C, Flodgren P, Maneval EC, Chen I, Vogelzang NJ (2009a) Phase II study of sunitinib administered in a continuous once-daily dosing regimen in patients with cytokine-refractory metastatic renal cell carcinoma. J Clin Oncl 27: 4068–4075
Escudier B, Szczylik C, Hutson TE, Demkow T, Staehler M, Rolland F, Negrier S, Laferriere N, Scheuring UJ, Cella D, Shah S, Bukowski RM (2009b) Randomized phase II trial of first-line treatment with sorafenib versus interferon Alfa-2a in patients with metastatic renal cell carcinoma. J Clin Oncol 27: 1280–1289
Escudier B, Szczylik C, Hutson TE, Demkow T, Staehler M, Rolland F, Negrier S, Laferriere N, Scheuring UJ, Cella D, Shah S, Bukowski RM (2009c) Randomized phase II trial of first-line treatment with sorafenib versus interferon Alfa-2a in patients with metastatic renal cell carcinoma. J Clin Oncol 27: 1280–1289
Flanigan RC, Salmon SE, Blumenstein BA, Bearman SI, Roy V, McGrath PC, Caton Jr JR, Munshi N, Crawford ED (2001) Nephrectomy followed by interferon alfa-2b compared with interferon alfa-2b alone for metastatic renal-cell cancer. N Engl J Med 345: 1655–1659
Gore ME, Szczylik C, Porta C, Bracarda S, Bjarnason GA, Oudard S, Hariharan S, Lee SH, Haanen J, Castellano D, Vrdoljak E, Schöffski P, Mainwaring P, Nieto A, Yuan J, Bukowski R (2009) Safety and efficacy of sunitinib for metastatic renal-cell carcinoma: an expanded-access trial. Lancet Oncol 10: 757–763
Gossage L, Eisen T (2010) Alterations in VHL as potential biomarkers in renal-cell carcinoma. Nat Rev Clin Oncol 7: 277–288
Hahn OM, Yang C, Medved M, Karczmar G, Kistner E, Karrison T, Manchen E, Mitchell M, Ratain MJ, Stadler WM (2008) Dynamic contrast-enhanced magnetic resonance imaging pharmacodynamic biomarker study of sorafenib in metastatic renal carcinoma. J Clin Oncol 26: 4572–4578
Hainsworth JD, Spigel DR, Burris III HA, Waterhouse D, Clark BL, Whorf R (2010) II Phase trial of bevacizumab and everolimus in patients with advanced renal cell carcinoma. J Clin Oncol 28: 2131–2136
Heng DY, Xie W, Regan MM, Warren MA, Golshayan AR, Sahi C, Eigl BJ, Ruether JD, Cheng T, North S, Venner P, Knox JJ, Chi KN, Kollmannsberger C, McDermott DF, Oh WK, Atkins MB, Bukowski RM, Rini BI, Choueiri TK (2009) Prognostic factors for overall survival in patients with metastatic renal cell carcinoma treated with vascular endothelial growth factor-targeted agents: results from a large, multicenter study. J Clin Oncol 27 (34): 5794–5799
Houk BE, Bello CL, Poland B, Rosen LS, Demetri GD, Motzer RJ (2009) Relationship between exposure to sunitinib and efficacy and tolerability endpoints in patients with cancer: results of a pharmacokinetic/pharmacodynamic meta-analysis. Cancer Chemother Pharmacol 66: 357–371
Hudes G, Carducci M, Tomczak P, Dutcher J, Figlin R, Kapoor A, Staroslawska E, Sosman J, McDermott D, Bodrogi I, Kovacevic Z, Lesovoy V, Schmidt-Wolf IG, Barbarash O, Gokmen E, O’Toole T, Lustgarten S, Moore L, Motzer RJ, Global ARCC Trial (2007) Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med 356: 2271–2278
Hutson TE, Davis ID, Machiels JP, De Souza PL, Rottey S, Hong BF, Epstein RJ, Baker KL, McCann L, Crofts T, Pandite L, Figlin RA (2010) Efficacy and safety of pazopanib in patients with metastatic renal cell carcinoma. J Clin Oncol 28: 475–480
Kontovinis LF, Papazisis KT, Touplikioti P, Andreadis C, Mouratidou D, Kortsaris AH (2009) Sunitinib treatment for patients with clear-cell metastatic renal cell carcinoma: clinical outcomes and plasma angiogenesis markers. BMC Cancer 9: 82
Motzer RJ, Bukowski RM, Figlin RA, Hutson TE, Michaelson MD, Kim ST, Baum CM, Kattan MW . Prognostic nomogram for sunitinib in patients with metastatic renal cell carcinoma. Cancer (2008a); 113: 1552–1558
Motzer RJ, Bukowski RM (2006) Targeted therapy for metastatic renal cell carcinoma. J Clin Oncol 24: 5601–5608
Motzer RJ, Escudier B, Oudard S, Hutson TE, Porta C, Bracarda S, Grünwald V, Thompson JA, Figlin RA, Hollaender N, Urbanowitz G, Berg WJ, Kay A, Lebwohl D, Ravaud A, RECORD-1 Study Group. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet (2008b); 372: 449–456
Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Oudard S, Negrier S, Szczylik C, Pili R, Bjarnason GA, Garcia-del-Muro X, Sosman JA, Solska E, Wilding G, Thompson JA, Kim ST, Chen I, Huang X, Figlin RA (2009) Overall survival and updated results for sunitinib compared with interferon alfa in patients with metastatic renal cell carcinoma. J Clin Oncol 27: 3584–3590
Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Rixe O, Oudard S, Negrier S, Szczylik C, Kim ST, Chen I, Bycott PW, Baum CM, Figlin RA (2007) Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med 356: 115–124
Na X, Wu G, Ryan CK, Schoen SR, di'Santagnese PA, Messing EM (2003) Overproduction of vascular endothelial growth factor related to von Hippel-Lindau tumor suppressor gene mutations and hypoxia-inducible factor-1 alpha expression in renal cell carcinomas. J Urol 170(2 Pt 1): 588–592
O’Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, Lane H, Hofmann F, Hicklin DJ, Ludwig DL, Baselga J, Rosen N (2006) mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res 66: 1500–1508
Oliner J, Min H, Leal J, Yu D, Rao S, You E, Tang X, Kim H, Meyer S, Han SJ, Hawkins N, Rosenfeld R, Davy E, Graham K, Jacobsen F, Stevenson S, Ho J, Chen Q, Hartmann T, Michaels M, Kelley M, Li L, Sitney K, Martin F, Sun JR, Zhang N, Lu J, Estrada J, Kumar R, Coxon A, Kaufman S, Pretorius J, Scully S, Cattley R, Payton M, Coats S, Nguyen L, Desilva B, Ndifor A, Hayward I, Radinsky R, Boone T, Kendall R (2004) Suppression of angiogenesis and tumor growth by selective inhibition of angiopoietin-2. Cancer Cell 6 (5): 507–516
Rini BI, Atkins MB (2009) Resistance to targeted therapy in renal-cell carcinoma. Lancet Oncol 10 (10): 992–1000
Rini BI, Halabi S, Rosenberg JE, Stadler WM, Vaena DA, Ou SS, Archer L, Atkins JN, Picus J, Czaykowski P, Dutcher J, Small EJ (2008a) Bevacizumab plus interferon alfa compared with interferon alfa monotherapy in patients with metastatic renal cell carcinoma: CALGB 90206. J Clin Oncol 26: 5422–5428
Rini BI, Michaelson MD, Rosenberg JE, Bukowski RM, Sosman JA, Stadler WM, Hutson TE, Margolin K, Harmon CS, DePrimo SE, Kim ST, Chen I, George DJ (2008b) Antitumor activity and biomarker analysis of sunitinib in patients with bevacizumab-refractory metastatic renal cell carcinoma. J Clin Oncol 26: 3743–3748
Rini BI, Wilding G, Hudes G, Stadler WM, Kim S, Tarazi J, Rosbrook B, Trask PC, Wood L, Dutcher JP (2009) Phase II study of axitinib in sorafenib-refractory metastatic renal cell carcinoma. J Clin Oncol 27: 4462–4468
Rixe O, Bukowski RM, Michaelson MD, Wilding G, Hudes GR, Bolte O, Motzer RJ, Bycott P, Liau KF, Freddo J, Trask PC, Kim S, Rini BI (2007) Axitinib treatment in patients with cytokine-refractory metastatic renal-cell cancer: a phase II study. Lancet Oncol 8: 975–984
Rixe O, Dutcher J, Motzer R, Wilding G, Stadler W, Garrett M, Pithavala Y, Kim S, Tarazi J, Rini B (2008) Association between diastolic blood pressure (DBP) G) ⩾90 mm Hg and efficacy in patients with metastatic renal cell carcinoma receiving axitinib (AG-013736). Ann Oncol 19 (suppl 8): viii189(abstr 587) PD 2009
Sablin MP, Negrier S, Ravaud A, Oudard S, Balleyguier C, Gautier J, Celier C, Medioni J, Escudier B (2009) Sequential sorafenib and sunitinib for renal cell carcinoma. J Urol 182: 29–34
Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM (2006) Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell 22: 159–168
Sternberg C, Szczylik E, Lee PV, Salman J, Mardiak ID, Davis L, Pandite M, Chen L, McCann L, Hawkins R (2009) A randomized, double-blind phase III study of pazopanib in treatment-naive and cytokine-pretreated patients with advanced renal cell carcinoma (RCC). J Clin Oncol 27(Suppl): 15s (abstract 5021)
Valent P (2008) Emerging stem cell concepts for imatinib-resistant chronic myeloid leukaemia: implications for the biology, management, and therapy of the disease. Br J Haematol 142: 361–378
van der Veldt AA, Meijerink MR, van den Eertwegh AJ, Haanen JB, Boven E (2010) Choi response criteria for early prediction of clinical outcome in patients with metastatic renal cell cancer treated with sunitinib. Br J Cancer 102: 803–809
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Powles, T., Chowdhury, S., Jones, R. et al. Sunitinib and other targeted therapies for renal cell carcinoma. Br J Cancer 104, 741–745 (2011). https://doi.org/10.1038/sj.bjc.6606061
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6606061
Keywords
This article is cited by
-
Phase 1 study of everolimus and low-dose oral cyclophosphamide in patients with metastatic renal cell carcinoma
Cancer Immunology, Immunotherapy (2019)
-
Phosphorylation of mTOR and S6RP predicts the efficacy of everolimus in patients with metastatic renal cell carcinoma
BMC Cancer (2014)
-
Sphingosine Kinase-1 Activation Causes Acquired Resistance Against Sunitinib in Renal Cell Carcinoma Cells
Cell Biochemistry and Biophysics (2014)
-
Prognostic implication of extrarenal metabolic tumor burden in advanced renal cell carcinoma treated with targeted therapy after nephrectomy
Annals of Nuclear Medicine (2013)
-
Incidence and risk of hypertension with pazopanib in patients with cancer: a meta-analysis
Cancer Chemotherapy and Pharmacology (2013)
