
Abstract
Background:
Sustained p38MAPK phosphorylation upregulates p75 neurotrophin (p75NTR) and induces apoptosis in Ewing's sarcoma family of tumours (ESFT). As fenretinide induces ESFT death through sustained p38MAPK phosphorylation, we hypothesised that this may be effected through upregulation of death receptors (DRs) and that treatment of fenretinide plus DR ligands may enhance apoptosis.
Methods:
DR expression was determined by flow cytometry. Trypan blue exclusion assays, caspase-8 flow cytometry and immunoblotting for Bid were used to measure cell death.
Results:
Fenretinide upregulated cell surface expression of tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) receptors, FAS and p75NTR, in an ASK1- and p38α-dependent manner. Cotreatment with fenretinide and DR ligands resulted in synergistic death compared with either agent alone; caspase-8 and Bid were cleaved in a time-dependent manner. Fenretinide did not increase DR expression in non-malignant cells. Furthermore, fenretinide, TRAIL or a combination of both agents was non-cytotoxic to non-malignant cells. Etoposide and actinomycin D increased expression of all DRs examined, whereas vincristine increased FAS alone. Only actinomycin D and TRAIL, and etoposide with TRAIL or FasL, enhanced death compared with either agent alone.
Conclusion:
The synergistic death observed with fenretinide and DR ligands suggests that this combination may be an attractive strategy for the treatment of ESFT.
Similar content being viewed by others


Main
Ewing's sarcoma family of tumours (ESFTs) arise in bone and soft tissue sites. ESFT occurs at all ages, although there is a peak incidence at 10–25 years of age. Recurrence and metastasis pose the greatest challenge for successful treatment of ESFT; patients in this cohort have a 20% or less 5-year disease-free survival rate (Proctor et al, 2009). There is therefore an urgent need for effective treatment strategies. Fenretinide, a synthetic retinamide with promising chemopreventive and chemotherapeutic properties, is well tolerated in both adult (Hail et al, 2006) and paediatric (Garaventa et al, 2003; Villablanca et al, 2006; Formelli et al, 2008) phase I clinical trials. We have previously demonstrated that fenretinide induces apoptosis through generation of reactive oxygen species (ROS) and phosphorylation of p38 mitogen-activated protein kinase (p38MAPK) in ESFT. Delayed growth of subcutaneous ESFT in nude mice was also observed following fenretinide treatment (Myatt et al, 2005).
The tumour necrosis factor (TNF) receptor (TNFR) superfamily includes a subclass of receptors known as death receptors (DRs), so called as they contain a cytoplasmic region called the death domain (DD) that is important for the induction of DR-mediated apoptosis. Members of the DR family include TNF-R1, p75 neurotrophin (p75NTR), FAS, DR4 and DR5 (Debatin and Krammer, 2004; Ashkenazi, 2008). Binding of TNF superfamily ligands to their cognate receptors induces receptor trimerisation and recruitment of Fas-associated death domain (FADD), which subsequently recruits procaspase-8 to form the death-inducing signalling complex (DISC). This association of proteins induces caspase-8 cleavage, which subsequently cleaves caspase-3 to induce a non-mitochondrial apoptotic pathway (Debatin and Krammer, 2004; Ashkenazi, 2008). Bid is a BH3-only member of the Bcl-2 family that is cleaved by caspase-8 in response to certain stimuli, whereby it translocates to the mitochondria to induce the oligomerisation of BAX or BAK and release of cytochrome C. In some cell types, sufficient quantities of caspase-8 are present to induce apoptosis independently of the mitochondria, whereas in cells with low levels of caspase-8, Bid cleavage is required to amplify death through mitochondria (Debatin and Krammer, 2004; Ashkenazi, 2008).
Tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) binds to DR4 and DR5, and to three decoy receptors (DcRs) that either lack an intracellular DD in the case of DcR1 or contain a truncated DD in the case of DcR2. The third DcR, osteoprotegrin is a soluble receptor. These DcRs do not induce apoptosis because of the absence of a functional DD (Ashkenazi, 2008). As TRAIL is reported to be selective in inducing apoptosis in malignant cells, although sparing healthy cells (Ashkenazi, 2008), numerous proapoptotic receptor agonists have been developed that include recombinant TRAIL and monoclonal agonistic antibodies against DR4 and DR5. Many show promise in clinical trials, both as monotherapies and in combination with biological agents, including histone deacetylase inhibitors (HDACi), bortezomib and chemotherapeutics (Ashkenazi, 2008).
Ewing's sarcoma family of tumour cells and tumour samples express both TRAIL DR and FAS (Kontny, 2006), although p75NTR has only been examined in cell lines (Westwood et al, 2002; Williamson et al, 2004). Approximately 80% of ESFT cell lines examined are reported to be sensitive to TRAIL-induced apoptosis (Kontny, 2006), and those that are resistant can be sensitised by pretreatment with interferon gamma (IFNγ; Kontny, 2006; Lissat et al, 2007). Enhanced death is also reported when ESFT cells are treated with TRAIL and bortezomib (Lu et al, 2008) or HDACi (Sonnemann et al, 2007). Recent analysis of the agonistic DR4 monoclonal antibody Mapatumumab in a paediatric preclinical testing program failed to demonstrate preclinical efficacy in subcutaneous ESFT models (Smith et al, 2009). However, the agonistic DR5 monoclonal antibody (M413; Amgen, Cambridge, UK) is reported to decrease ESFT growth following intramuscular injection (Merchant et al, 2004). Furthermore, delivery of TRAIL through non-viral gene therapy diminished tumour growth and increased animal survival in ESFT (A673) mouse xenografts (Picarda et al, 2010). A phase I trial is currently recruiting young patients with solid tumours or lymphomas refractory to conventional chemotherapeutics to assess the effects of the DR5 agonistic antibody Lexatumumab either alone or in combination with IFNγ (ClinicalTrials.gov Identifier: NCT00428272).
We previously demonstrated that sustained p38MAPK phosphorylation is required for the upregulation of p75NTR protein expression and induction of apoptosis by basic fibroblast growth factor (bFGF; Williamson et al, 2004). As fenretinide also induces ESFT apoptosis through sustained p38MAPK phosphorylation (Myatt et al, 2005), we hypothesised that fenretinide-induced death may be effected in part through the induction of DR. If correct, we hypothesised that treatment with DR ligands plus fenretinide should amplify the initial apoptosis induced by fenretinide. Results indicate that fenretinide induced upregulation of DR at the cell surface in an apoptosis signal-regulating kinase (ASK) 1- and p38α-dependent manner. Furthermore, cotreatment with DR ligands and fenretinide resulted in synergistic death compared with either agent alone. However, only certain chemotherapeutic drugs (etoposide and actinomycin D) upregulated DR and this did not always occur in a p38MAPK-dependent manner or correspond with enhanced cell death when cotreated with drug and DR ligands.
Materials and methods
Cells and reagents
The cell lines used are described in Supplementary Table 1. All reagents were prepared and stored according to the manufacturer's instructions and obtained from the following suppliers: recombinant TRAIL and Fas ligand (FasL) from R&D systems (Abingdon, UK), γ-32P-ATP from GE Healthcare Life Sciences (Little Chalfont, UK). All other chemicals were obtained from Sigma (Poole, UK) unless stated otherwise. Fenretinide (gift from the National Cancer Institute), BAY 11-70892 (Calbiochem, Nottingham, UK) and SB202190 (Calbiochem) were prepared as previously described (Myatt et al, 2005; White and Burchill, 2008). BIRB0796 (10 mM stock in DMSO, stored at −20 °C) was purchased from Dr Hilary McLaughlan, University of Dundee. All antibody dilutions were determined empirically. Total and phosphorylation-specific p38MAPK antibodies, used at a dilution of 1 : 5000 and 1 : 10000, respectively, anti-MKK3 and anti-phospho-MKK3/6 (this antibody recognises a phosphorylated activation loop conserved between MKK3 and MKK6) antibodies, used at 1 : 500, and Bid antibody (1 : 500) were all purchased from Cell Signaling Technology (Danvers, MA, USA). Protein A/G PLUS-Agarose, antitubulin (1 : 5000) and anti-ASK 1 (H-300, 1 : 100) antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Antibodies for flow cytometry were diluted in FACS buffer (PBS, 1% fetal calf serum (SeraLab, Haywards Heath, UK), 0.1% sodium azide). FAS-FITC (Beckman Coulter, High Wycombe, UK) and IgG1-FITC (Dako Cytomation, Ely, UK) antibodies were used at 1 : 10 dilution. p75NTR antibody (Promega, Southampton, UK) was used at 1 : 500. All TRAIL-R antibodies (Alexis Biochemicals, Exeter, UK) were used at 1 : 100 dilutions, except for DR4 (1 : 50). Secondary antibodies goat F(ab2) antirabbit IgG-PE (Caltag Laboratories, Paisley, UK) and goat F(ab)2 antimouse IgG-PE (Southern Biotechnology Associates from Cambridge Biosciences, Cambridge, UK) were used at 1 : 200.
Cell surface expression of receptors
Cells were harvested, washed in FACS buffer and incubated with antibodies (4 °C, 30 min for both primary and secondary antibodies). Controls included secondary antibody only (TRAIL receptors and p75NTR) or IgG1-FITC antibody (FAS). Cells were washed, resuspended in 300 μl FACS buffer and cell surface expression was determined by flow cytometry (FACSCalibur, BD, Oxford, UK). Data were analysed using CellQuest software (BD Biosciences, Oxford, UK). Results are presented as (mean of the median fluorescence intensity for each DR minus median fluorescence with secondary (TRAIL and p75NTR) or IgG1-FITC (FAS) antibody)±s.e.m. (n=9). The following cell lines and treatments were used as positive controls to optimise antibodies (data not shown): jurkat cells (DR5), A375 cells (DR4), MCF-7 cells (DcR2), doxorubicin (500 ngml−1, 15 h)-treated MCF-7 cells (DcR1 and FAS) and bFGF (20 ngml−1, 16 h)-treated TC-32 cells (p75NTR).
Fenretinide array
RNA was extracted from both untreated and fenretinide (3 μ M, 8 h)-treated TC-32 cells using Ultraspec (Biotecx, Houston, TX, USA), as previously described (Brownhill et al, 2007). Total RNA (2 μg) was used to generate a biotin-labelled apoptosis-specific cDNA library, which was hybridised with a GEArray Q-series human apoptosis array (SA Biosciences, Frederick, MD, USA), according to the manufacturer's instructions. Arrays were processed for chemiluminescence, data were extracted with ScanAlyze software (Eisen Lab, Berkeley, CA, USA) and analysed using the GEArray analyser software (SA Biosciences).
Electroporation of TC-32 cells with small interfering RNA (siRNA)
Cells were electroporated as described previously (Myatt et al, 2005; White and Burchill, 2008) with 500 nM siRNA directed against ASK1 (Santa Cruz Biotechnology; a pool of three individual ASK1 siRNAs) or scrambled siRNA (Silencer Negative Control #1; Ambion, Huntington, UK). Sequences of all siRNAs are company propriety. ASK1 knockdown was confirmed by quantitative real-time RT–PCR using ASK1 Taqman Gene Expression Assay ID: Hs00178726 (Applied Biosystems, Warrington, UK) and β2 M probes and primers, as described previously (Brownhill et al, 2007). Electroporated cells were analysed after 48 h for receptor expression by flow cytometry or viable cell number using the Trypan blue exclusion assay.
ASK 1 immunecomplex kinase assay
Fenretinide (3 μ M, 0–60 min)-treated cells were lysed in buffer (20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerol phosphate, 1 mM Na3VO4, 1 μg/ml leupeptin and 1 mM phenylmethylsulphonyl fluoride) on ice for 5 min. Lysates were clarified by centrifugation (10 000 g, 15 min, 4 °C) and protein concentration was determined using the Bio-Rad DC protein assay (Bio-Rad Laboratories, Hemel Hempstead, UK). Total protein lysate (200 μg) was incubated with anti-ASK1 antibody (2 μg, 1 h), and then incubated with 20 μl protein A/G-sepharose overnight at 4 °C with rotation. Beads were collected by centrifugation (4000 g, 3 min, 4 °C) and washed twice with 500 μl lysis buffer and kinase buffer (25 mM Tris (pH 7.5), 5 mM β-glycerolphosphate, 2 mM DTT, 0.1 mM Na3VO4 and 10 mM MgCl2). Pellets were resuspended in 50 μl kinase buffer containing 200 μ M ATP, 2 μCi γ-32P-ATP and 4 μg myelin basic protein (MBP), and incubated at 30 °C for 30 min. Reactions were terminated by adding 25 μl 2 × SDS loading buffer. Samples were heated at 95 °C for 5 min and separated by SDS–PAGE. The gel was rinsed in water, stained in Bio-safe Commassie (Bio-Rad Laboratories) for 1 h and dried under vacuum. ASK1 activity was assessed by incorporated 32P in MBP, as determined by autoradiography, and quantified using phosphorimager (Quantity One software, Bio-Rad Laboratories). SH-SY5Y cells treated with or without 6-hydroxydopamine (6-OHDA; 100 μ M, 1 h) served as positive and negative controls, respectively.
Cell viability
Viable cell number was determined using the Vi-cell Trypan blue exclusion assay (Beckman Coulter) as described previously (White and Burchill, 2008).
Caspase-8 activity
Caspase-8 activation was determined using the fluorochrome inhibitor of caspases (FLICA) FAM-LETD-FMK apoptosis detection kit (Immunochemistry Technologies, Bloomington, MN, USA) according to the manufacturer's instructions. The FLICA inhibitor covalently binds to a reactive cysteine residue on the large subunit of active caspase-8 heterodimer. This inhibits any further caspase activity and retains FLICA intracellularly so that active caspase-8 molecules can be quantified by flow cytometry. Jurkat cells treated with or without etoposide (30 μ M, 16 h) served as positive and negative controls, respectively.
Immunoblotting
Lysates were extracted and proteins detected by immunoblotting and visualised using the Odyssey infrared imaging system (Li-cor, Lincoln, NE, USA) as described previously (Myatt et al, 2005).
Statistical analyses
Statistical analyses were undertaken using GraphPad Prism 5 or SAS 9. Data were analysed by analysis of variance and a Bonferroni post hoc test when comparing three or more conditions. An interaction effect between drug and DR ligand was assessed to determine whether enhanced death had occurred. An unpaired two-tailed t-test was used to analyse differences in untreated versus fenretinide-treated non-malignant cells. Variations between means were considered significantly different at P⩽0.05.
Results
Fenretinide upregulates DR and DcR in ESFT cells
Basal cell surface expression of TRAIL receptors, FAS and p75NTR, was established in a panel of ESFT cell lines. DR5 was the most abundantly expressed receptor in all cell types, whereas DR4 was expressed at much lower levels (Figure 1A). IgG1-FITC antibody was moderately expressed in all cells, except in SK-N-MC and TTC-466, with the highest expression detected in TC-32 cells. DcR1, DcR2 and p75NTR were marginally expressed in ESFT cells. RD-ES, SKES-1 and TC-32 were selected for further studies because of their heterogeneity of receptor expression and the genotypic differences described previously (Westwood et al, 2002; Brownhill et al, 2007). Neither DR nor DcR were expressed in the non-malignant primary normal human urothelial cells (NHUC) or mesenchymal stem cells (MSC, data not shown).

Fenretinide upregulates DR and DcR in ESFT cells. (A) Basal cell surface expression of DcR and DR was determined by antibody labelling cells and by analysing levels by flow cytometry. Results are presented as the mean of the median fluorescence intensity for each DR minus median fluorescence with secondary (TRAIL and p75NTR) or IgG1-FITC (FAS) antibody±s.e.m. (n=9). (B) ESFT cells were treated with fenretinide (3 μ M, 0–24 h) or vehicle control for 0, 6, 16 or 24 h and (C) NHUC and MSC were treated with fenretinide (3 μ M) for 24 h. Receptor cell surface expression was examined by antibody labelling and flow cytometry. Results are presented as the fold increase of the mean of the receptor median fluorescence intensity relative to untreated control samples±s.e.m. (n=6). *P⩽0.001, **P⩽0.05.
We previously demonstrated that bFGF-induced apoptosis is dependent on p38MAPK-mediated induction of p75NTR in ESFT cells (Westwood et al, 2002; Williamson et al, 2004). As fenretinide-induced apoptosis also requires sustained p38MAPK phosphorylation (Myatt et al, 2005), we hypothesised that DR upregulation may be involved in fenretinide-induced death. Fenretinide (3 μ M, 8 h) strongly upregulated DR5 cDNA and moderately increased DR4 and DcR2 cDNA in TC-32 cells (Supplementary Figure 1 and Supplementary Table 2). No changes in the expression of other DR pathway components (DcR1, FAS, caspase-8, caspase-10, TRAIL, FasL and FADD) were observed.
The effect of fenretinide on cell surface receptor expression was also examined. Fenretinide induced rapid upregulation of both DcRs in all ESFT cells (Figure 1B, four- to five-fold in TC-32 cells, two- to four-fold in RD-ES and SKES-1 cells, P⩽0.001). DR4 was upregulated in a time-dependent manner in RD-ES and SKES-1 cells, and was strongly upregulated in TC-32 cells at 6 h, with no further increase observed up to 24 h. DR5 was increased at 16 h in SKES-1 cells and at 24 h in RD-ES and TC-32 cells (P⩽0.01). Fenretinide increased FAS expression from 16 h in TC-32 cells (P⩽0.01) and at 24 h in SKES-1 cells (P⩽0.05) but had no effect on FAS expression in RD-ES cells. p75NTR expression was maximally increased in RD-ES cells at 16 h (4.9-fold), whereas a time-dependent increase occurred in SKES-1 and TC-32 cells (2.7-fold and 2.8-fold, respectively, at 24 h, P⩽0.001). Fenretinide (3 μ M, 24 h) did not affect DR4, DR5 or p75NTR expression in NHUC (Figure 1C). However, it did induce a 2.5-fold downregulation of FAS and upregulation of both DcR1 (4.4-fold) and DcR2 (4.7-fold, P⩽0.001). Fenretinide had no effect on receptor expression in MSC, suggesting that fenretinide-induced DR upregulation is selective to cancer cells.
Fenretinide-induced upregulation of DR is dependent on phosphorylation of ASK1 and p38α
We examined the hypothesis that fenretinide-induced DR upregulation may be effected through p38MAPK using the pharmacological inhibitor SB202190. SB202190 (20 μ M, 1 h) and fenretinide cotreatment (3 μ M, 24 h) decreased cell surface expression of DR5 in SKES-1 and TC-32 cells, of FAS in TC-32 cells and of p75NTR in all cell types, compared with fenretinide-induced DR upregulation (Figure 2A, P⩽0.05: p75NTR in TC-32 cells, P⩽0.001: other receptors and cell types). SB202190 alone had no effect on DR expression. These observations suggest that upregulation of some DRs by fenretinide may occur in a p38MAPK-dependent manner.


Fenretinide upregulation of DR is dependent on phosphorylation of ASK1, and p38α. Ewing's sarcoma family of tumour cells were pretreated with (A) SB202190 (20 μ M, 1 h) or (B) BIRB0796 (0.1 or 1 μ M, 24 h) before fenretinide treatment (3 μ M, 24 h) and DR expression was determined by antibody labelling and flow cytometry. Results are presented as the fold increase of the mean of the receptor median fluorescence intensity relative to untreated control samples±s.e.m. (n=6). (Ci) TC-32 cells were electroporated with scrambled or ASK1 siRNA (500 nM), and ASK1 kinase activity was assessed 48 h later by immunecomplex kinase assays using MBP as the substrate. Samples were subsequently resolved by SDS–PAGE and gels were analysed by quantitative autoradiography using a PhosphorImager system. Untreated and 6-OHDA (100 μ M, 1 h)-treated SH-SY5Y cells served as negative and positive controls, respectively, for ASK1 kinase activity. Graph shows ASK1 activity calculated as fold increase over untreated scrambled siRNA cells. (Cii) TC-32 cells were pretreated with vitamin C (100 μ M, 1 h) before fenretinide treatment (3 μ M, 10 min) and ASK1 immunecomplex kinase assays were performed. (Ciii) TC-32 cells were electroporated with scrambled or ASK1 siRNA (500 nM). Protein expression was determined 48 h later by immunoblot analysis for total p38MAPK and phospho-p38MAPK and total MKK3 and phospho-MKK3/6. Anisomycin (25 μg/ml, 30 min)-treated TC-32 cells served as a positive control for MKK3/6 phosphorylation. Equal protein loading was confirmed by hybridisation to tubulin. M=molecular weight markers. Representative images from three independent experiments are shown. (D) TC-32 cells were electroporated with scrambled or ASK1 siRNA (500 nM) and were treated with fenretinide (3 μ M, 24 h) 24 h after electroporation. DR expression was determined by antibody labelling and flow cytometry, and results presented as the fold increase of the mean of the receptor median fluorescence intensity relative to untreated control samples±s.e.m. (n=6). *P⩽0.001, **P⩽0.01, ***P⩽0.05.
BIRB0796 was used to delineate which p38MAPK isoform was required for fenretinide-induced DR upregulation; BIRB0796 inhibits p38α at 0.1 μ M, whereas 1 μ M inhibits all four isoforms (Kuma et al, 2005). BIRB0796 pretreatment (0.1 or 1 μ M, 24 h) before fenretinide treatment (3 μ M, 24 h) suppressed fenretinide-induced upregulation of DR5, p75NTR and FAS in all cell types (Figure 2B, P⩽0.001). Although BIRB0796 treatment alone had no effect on DR expression, BIRB0796 and fenretinide cotreatment significantly decreased basal FAS expression in RD-ES cells. Furthermore, no significant difference was observed between the effects of 0.1 and 1 μ M of BIRB pretreatment, indicating that p38α is the isoform most likely to be required for fenretinide-induced DR upregulation.
Apoptosis signal-regulating kinase 1 is a redox-regulated MAPK kinase that regulates stress-activated MAPK in response to apoptotic stimuli (Coulthard et al, 2009). The effect of fenretinide on ASK1 kinase activity was determined by immunecomplex kinase assays using MBP as a substrate. Preliminary data indicated that in all ESFT cell lines examined (RD-ES, SKES-1 and TC-32), fenretinide (3 μ M) induced ASK1 kinase activity from 5 min, with maximium activation observed at 10 min. MBP phosphorylation decreased with time but remained above basal levels for up to 60 min (data not shown). To confirm whether ASK1 was an upstream regulator of p38MAPK-dependent fenretinide-induced DR upregulation, siRNA was used. ASK1 siRNA (500 nM, 48 h) reduced ASK1 mRNA levels by 79% in TC-32 cells compared with scrambled control cells (Supplementary Figure 2). Fenretinide (3 μ M) induced ASK1 kinase activity in scrambled siRNA-treated TC-32 cells and MBP phosphorylation was abolished in ASK1 siRNA-treated TC-32 cells (Figure 2Ci). Phosphorylation was comparable to that detected in the positive control SHSY-5Y neuroblastoma cells treated with 6-OHDA (100 μ M, 1 h). Furthermore, no ASK1 activity or p38MAPK phosphorylation was detected in SHEP1 neuroblastoma cells (data not shown), a cell line that we have previously demonstrated to be resistant to fenretinide (Myatt et al, 2005; White and Burchill, 2008). Pretreatment of TC-32 cells with vitamin C (100 μ M, 1 h) before fenretinide treatment (3 μ M, 10 min) completely abolished ASK1 activity (Figure 2Cii; data not shown for SKES-1 and RD-ES cells), suggesting that fenretinide-induced ROS production is upstream of ASK1 activity in ESFT cells.
Immunoblot analysis identified fenretinide-induced p38MAPK phosphorylation in scrambled siRNA-treated ESFT cells at 30 min. However, lower phosphorylation levels were observed in ASK1 siRNA-treated cells in response to fenretinide (Figure 2Ciii: representative image shown for TC-32 cells, data not shown for SKES-1 and RD-ES cells), thus confirming that p38MAPK is downstream of ASK1 in the fenretinide-induced apoptotic pathway. A similar experimental design was used to identify the intermediate regulatory MAPK kinase (MKK3 or MKK6; Coulthard et al, 2009) between ASK1 and p38MAPK in the fenretinide-induced apoptotic pathway. MKK3/6 was robustly phosphorylated 30–60 min after fenretinide treatment in scrambled siRNA-treated ESFT cells (Figure 2Ciii; representative image shown for TC-32 cells, data not shown for SKES-1 and RD-ES cells). This was a comparable level of phosphorylation to that detected in anisomycin-treated (25 μg, 30 min) TC-32 cells used as a positive control. Lower levels of MKK3/6 phosphorylation were detected in ASK1-treated siRNA cells, suggesting that these MAPKK are downstream of ASK1 in the fenretinide-induced apoptotic pathway. No changes in total MKK3 or MKK6 expression were observed in either cell type.
Fenretinide induced DR upregulation in scrambled siRNA-treated TC-32 cells (Figure 2D), consistent with previous observations in unelectroporated cells (Figure 1B). However, reduced DR levels were observed in ASK1 siRNA-treated cells compared with scrambled siRNA cells, indicating that ASK1 is required for fenretinide-induced upregulation of DR (P⩽0.001: DR5 and p75NTR, P⩽0.01: FAS). Furthermore, ASK1 siRNA rescued fenretinide-induced death by 52% compared with scrambled siRNA cells (P⩽0.01, Supplementary Figure 3), demonstrating that fenretinide induces DR upregulation and cell death in an ASK1-p38MAPK-dependent manner in ESFT cells.
Fenretinide sensitises ESFT cells to the DR apoptotic pathway to enhance cell death
As increased cell surface expression of DR would increase receptor availability to cognate ligands, we hypothesised that cotreatment of ESFT cells with fenretinide plus DR ligands (TRAIL for DR5, nerve growth factor (NGF) for p75NTR and FasL for FAS) may enhance death. Treatment with DR ligands (0–40 ng ml−1, 24 h) alone did not decrease viable cell number (Figure 3A; we have performed viability assays with up to 400 ngml−1 TRAIL in six ESFT cell lines and have not observed cell death, unpublished observation). However, the combination of fenretinide and DR ligands resulted in synergistic cell death (reductions were 43% for TRAIL and NGF and 36% for FasL with 40 ng ml−1 DR ligand and 1.5μ M fenretinide) compared to fenretinide alone (Figure 3A, statistical interactions P⩽0.001) in TC-32 cells. Furthermore, neither DR ligands (40 ng ml−1, 24 h) nor fenretinide (3 μ M, 16 h) or a combination of both agents had any effect on viable cell number in non-malignant NHUC or MSC (data not shown; mean viable cell number calculated as percentage of untreated control ranged from 95±4% to 107±4%).

Fenretinide sensitises ESFT cells to the DR apoptotic pathway to enhance cell death. (A) TC-32 cells were treated with fenretinide (Fen; 1.5 μ M, 16 h) or vehicle control (Veh; ethanol treated cells), media were changed and cells were subsequently treated with the DR ligands TRAIL, FasL or NGF (0–40 ng/ml, 24 h). Viable cell number was determined by the Trypan blue exclusion assay. Results are presented as the mean of the viable cell number calculated as a percentage of untreated control cells±s.e.m. (n=9). Statistics indicate significant interactions between fenretinide and DR ligands, which is indicative of enhanced cell death; *P⩽0.001. TC-32 cells were pretreated with fenretinide (1.5 μ M, 16 h) before DR ligands (40 ng/ml, 0-24 h) and caspase-8 activity was detected by flow cytometry. (Bi) Effect of fenretinide and (Bi) TRAIL, (Bii) FasL or NGF time course of caspase-8 cleavage in TC-32 cells. Results are presented as the mean of the percentage of cells with cleaved caspase-8, as detected within the M1 region±s.e.m. (n=9), *P⩽0.001. Untreated and etoposide (30 μ M, 24 h)-treated Jurkat cells served as negative and positive control samples, respectively. (C) Total protein lysates from ESFT cells pretreated with fenretinide (Fen, 1.5 μ M, 16 h) before TRAIL treatment (40 ng/ml, 24 h) were examined for both full length (FL, 22 kDa) and truncated (cleaved) bid (tBid, 15 kDa) by immunoblotting. Equal protein loading was confirmed by hybridisation to tubulin. M=molecular weight markers. Negative (Neg) control=untreated Jurkat cells, positive (Pos) control=etoposide-treated (25 μ M, 16 h) Jurkat cells. Immunoblots are representative from three independent experiments.
We next examined whether this enhanced death was effected through caspase-8 cleavage. No significant cleavage was observed following single treatments with fenretinide or DR ligands (Figure 3B and Supplementary Figure 4), supporting the hypothesis that fenretinide-induced apoptosis is effected through the mitochondrial death cascade (Myatt et al, 2005), and consistent with the observation that DR ligands do not reduce TC-32 viable cell number. However, combined treatment of fenretinide and TRAIL increased caspase-8 activity in a time-dependent manner, with 68% activity observed at 24 h (Figure 3Bi and Supplementary Figure 4, P⩽0.001). FasL and fenretinide induced 31% caspase-8 activation within 14 h exposure, which did not further increase up to 24 h, whereas NGF and fenretinide induced caspase-8 activity at 12 h (31%), with a further increase observed at 24 h (Figure 3Bii, P⩽0.001).
Immunoblot analysis demonstrated that fenretinide and TRAIL induced cleavage of Bid to generate a truncated form (tBid; Figure 3C) compared with untreated cells and cells treated with either agent alone. This was observed in all ESFT cell lines examined; cleavage was comparable to that in etoposide (25 μ M, 16 h)-treated Jurkat cells (positive control). These data indicate a putative role of truncated Bid in amplifying the DR pathway of apoptosis through the mitochondria in ESFT cells.
Specific chemotherapeutics upregulate DR and enhance ESFT cell death
Chemotherapeutics commonly used in the treatment of ESFT were also examined to determine whether they upregulated DR cell surface expression and enhanced DR ligand-induced cell death or whether this was a fenretinide-specific phenomenon. Etoposide increased expression of all DRs (Figure 4A, P⩽0.001), actinomycin D increased DR5 (P<0.001) and p75NTR (P=0.03), whereas vincristine only upregulated FAS (P⩽0.001). On using the calculated IC50, doxorubicin did not significantly increase DR expression (Figure 4A). However, at 10 times the doxorubicin IC50 value, upregulation of all DRs was observed (data not shown, P=0.002). An increase in p75NTR cell surface expression was detected when cells were treated with 10 times the vincristine IC50 (data not shown, P=0.001).

Specific chemotherapeutics upregulate DR and enhance ESFT cell death. (A) TC-32 cells were treated with the indicated drug IC50 or vehicle control for 48 h and cell surface DR expression was determined by antibody labelling and flow cytometry. Results are presented as the fold increase of the mean of the receptor median fluorescence intensity relative to untreated control samples±s.e.m. (n=6). *P<0.001, **P=0.03. (B) TC-32 cells were treated with chemotherapeutic agents (drug IC50, 48 h) or vehicle control, and ASK1 immunecomplex kinase assays were performed as described in figure legend 2C. Positive control=6-OHDA (100 μ M, 1 h)-treated SH-SY5Y cells. Graph depicts ASK1 kinase activity presented as fold increase of drug-treated samples over untreated control, as quantified by phosphorimager analysis. (C) TC-32 cells were treated with chemotherapeutic agents (drug IC50, 24 h) or vehicle control, media were changed and cells were subsequently treated with DR ligands (0–40 ng/ml, 24 h). Viable cell number was determined by the Trypan blue exclusion assay. Results are presented as the mean of the viable cell number calculated as a percentage of untreated control cells±s.e.m. (n=9). Statistics indicate significant interactions between drugs and DR ligands, which is indicative of enhanced cell death. *P⩽0.001, **P⩽0.01, ***P⩽0.05.
It is reported in the literature that etoposide induces DR upregulation through NF-κB transcriptional upregulation (Gibson et al, 2000; Shetty et al, 2002; Woo et al, 2004; Mendoza et al, 2008). BAY 11-70892, an inhibitor of IκBα phosphorylation and subsequent NF-κB translocation to the nucleus, was used to elucidate whether this was the mechanism of upregulation in ESFT cells. BAY 11-7082 had no effect on DR upregulation (data not shown), suggesting that NF-κB does not regulate etoposide-induced DR expression in ESFT cells. However, SB202190 pretreatment decreased both actinomycin D and etoposide-induced DR5 expression and actinomycin D-induced p75NTR expression (data not shown). Furthermore, ASK1 kinase activity increased after treatment with etoposide and vincristine but not after treatment with doxorubicin or actinomycin D (Figure 4B). Actinomycin D failed to activate ASK1, although SB202190 prevented DR5 and p75NTR upregulation, suggesting an alternative mechanism of upregulation. Collectively, these data are consistent with the hypothesis that both ASK1 and p38MAPK are required for the chemotherapeutic-induced upregulation of certain DRs in some ESFT cells.
TC-32 cells were pretreated with chemotherapeutics which significantly increased DR expression at the cell surface, and subsequently treated with DR ligands. Both TRAIL and FasL were dependent on the presence of etoposide to induce cell death, as neither ligand induced death when used alone (Figure 4C, P⩽0.05). No statistical interaction was observed between etoposide and NGF combined treatment, despite a 15% decrease in viable cell number. Actinomycin D cotreatment with TRAIL (significant interaction of P⩽0.001), but not with NGF, resulted in additive cell death, whereas vincristine and FasL cotreatment did not further decrease viable cell number compared with treatment with vincristine alone (Figure 4C). The observations with cotreatments of FasL and vincristine, and NGF with etoposide or actinomycin D, suggest that high levels of DR expression and treatment with the cognate ligand might not always correlate with increased cytotoxicity. The effect of ligands and doxorubicin was therefore examined because no significant increase in DR expression was observed with this drug. No enhanced death was observed with doxorubicin and with either TRAIL or NGF compared with doxorubicin alone (Figure 4C).
Discussion
In this study, we demonstrate for the first time that cotreatment with fenretinide and DR ligands enhanced ESFT cell death compared with fenretinide alone, through the induction of both the mitochondrial and DR apoptotic pathways (Figure 5); DR ligands alone did not induce death. Fenretinide upregulated cell surface expression of DRs TRAIL, FAS and p75NTR in an ASK1- and p38α-dependent manner. This is likely to be initiated through increased levels of ROS, as fenretinide-induced death (Myatt et al, 2005) and ASK1 phosphorylation are inhibited by antioxidants. ROS have previously been implicated in DR upregulation at the cell surface and in subsequent apoptosis in response to hydrogen peroxide (Kwon et al, 2008), 15-deoxy-delta12,14-prostaglandin J2 (Su et al, 2008), zerumbone (Yodkeeree et al, 2009) and arsenic trioxide (Woo et al, 2004), consistent with the hypothesis that induction of DR expression is effected through the generation of ROS. Induction of DR4 protein expression by bortezomib (Nakamura et al, 2007) and downregulation of DR5 cell surface expression by HDACi (Sonnemann et al, 2007) have previously been reported in ESFT cells.

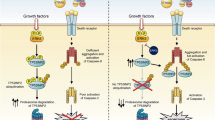
Combined treatment of fenretinide, etoposide or actinomycin D with TRAIL results in enhanced cell death. Treatment of ESFT cells with fenretinide leads to mitochondrial-dependent apoptosis through ROS generation, and ASK1-induced phosphorylation of p38MAPK. Fenretinide treatment also results in ASK1- and p38α-mediated upregulation of DR at the cell surface. Etoposide and actinomycin D had similar effects. Treatment with ligands to the cognate DR resulted in enhanced cell death, which was mediated through both caspase-8 and Bid cleavage (tBid=truncated Bid).
p38MAPK induces p75NTR protein expression in ESFT cells in response to bFGF (Williamson et al, 2004) and p75NTR mRNA and protein expression in prostate cancer cells by inflammatory agents (Quann et al, 2007; Khwaja et al, 2008). Furthermore, zerumbone induces DR4 but not DR5 protein expression through a p38MAPK-dependent mechanism in colon cancer cells (Yodkeeree et al, 2009). However, the p38MAPK isoform involved in these processes has not been identified. This is likely to be important for therapeutic interventions exploiting p38MAPK, as four isoforms exist that regulate varied biological processes (Coulthard et al, 2009). Induction of death through specific p38MAPK isoforms that do not result in unacceptable side effects and toxicities will be more attractive than if isoforms that are implicated in toxicity are involved. The mechanism by which ASK1 and p38MAPK upregulate DR in ESFT cells remains to be elucidated. Upregulation of both mRNA and cell surface expression of TRAIL DR by agents such as etoposide, proteasome inhibitors and arsenic trioxide is frequently reported to be mediated by p53 (Chen et al, 2008) and/or NF-κB (Woo et al, 2004; Chen et al, 2008; Mendoza et al, 2008; Song et al, 2008). However, we have found that DR upregulation in ESFT cells is not dependent on EWS–ETS fusion type, p53, p16 (Brownhill et al, 2007) or NF-κB status, suggesting that the manner of DR upregulation is cell type and stimulus dependent. DR5 is reported to be upregulated by the transcription factor CHOP in several cell lines by different stimuli, for example, 15-deoxy-delta12, 14-prostaglandin J2 (Su et al, 2008), proteosome inhibitors (Hetschko et al, 2008) and endoplasmic reticulum stress (Yamaguchi and Wang, 2004). One report has shown that fenretinide and TRAIL enhance apoptosis through CHOP-dependent DR5 upregulation (Kouhara et al, 2007). No differences in DR4 and FAS mRNA levels were reported; hence studies on these receptors were discontinued. These observations are consistent with our cDNA array data but importantly we discovered that fenretinide upregulated these receptors at the cell surface. Further investigation into how p38α upregulates DR at the surface of ESFT cells is required. This may be mediated by CHOP because it has previously been implicated in fenretinide-induced death (Hail et al, 2006) and is a known p38MAPK substrate (Coulthard et al, 2009).
p38MAPK inhibitors failed to reduce fenretinide-induced DR expression to basal levels, suggesting that other factors may also upregulate DR. c-Jun N-terminal kinase is an additional ASK1 substrate that is activated by fenretinide in certain cell types (Kang et al, 2008; Appierto et al, 2009). Several groups report dual activation of p38MAPK and c-Jun N-terminal kinase (Osone et al, 2004; Kim et al, 2006; Woo et al, 2009), suggesting that both MAPK may be required for fenretinide-induced DR upregulation in ESFT cells.
Cotreatment with fenretinide and DR ligands resulted in synergistic cell death through caspase-8 and Bid-dependent pathways, although confirmation that these are effectors of cell death requires validation through siRNA or inhibitor studies. This amplified death above that induced by either agent alone reflected activation through both the extrinsic (DR; Ashkenazi, 2008) and intrinsic (fenretinide; Myatt et al, 2005) apoptotic pathways (Figure 5). Fenretinide is also reported to activate caspase-8 and caspase-9 in glioblastoma (Das et al, 2008), meningioma and ovarian carcinoma (Cuello et al, 2004; Hail et al, 2006). We also demonstrated that etoposide and vincristine induce DR upregulation at the cell surface through ASK1 and p38MAPK, which subsequently results in enhanced death when cells are treated with DR ligands (although these effects were more modest than those observed with fenretinide). These observations support previously published data that etoposide increases DR5 mRNA and protein expression (Gibson et al, 2000), although they contradict studies in which vincristine had no effect on FAS mRNA expression in HL-60 leukaemic cells (Thomadaki et al, 2009) and the synergistic effect of actinomycin D through downregulation of XIAP with no effect on TRAIL DR expression (Ng et al, 2002). Doxorubicin has also been shown to induce DR5 cell surface expression in other cell types (Yoshida et al, 2003), suggesting that the effects of chemotherapeutics on DR expression is cell-line dependent. However, only actinomycin D, TRAIL and etoposide, with either TRAIL or FasL, enhanced cell death compared with either agent alone, consistent with the observation that high levels of DR expression do not necessarily correlate with ligand-induced cytotoxicity (Georgakis et al, 2005). In all drug combinations, a 20–40% cell population remained refractory to treatment. This may be explained by the presence of cancer stem cell like cells which are hypothesised to be responsible for resistance to therapy and tumour relapse (Hemmings, 2010).
Importantly, fenretinide did not increase DR expression in non-malignant MSC and NHUC. Furthermore, fenretinide, TRAIL or the combination of both agents was not cytotoxic to either of these non-malignant cell lines, indicating that the combination of fenretinide and TRAIL may have minimal toxicity. Interestingly, FAS was downregulated by fenretinide, and both DcRs were strongly increased in NHUC. We also observed that fenretinide upregulated TRAIL DcR in ESFT cells, which supports previous data in which TRAIL DcRs are upregulated alongside DR by apoptotic stimuli such as UV (Maeda et al, 2001), doxorubicin (Yoshida et al, 2003) and oxaplatin (Toscano et al, 2008). Collectively, these observations suggest a possible mechanism to evade apoptosis, the higher DcR to DR ratio being likely to drive cells towards a survival pathway. Cytoprotective roles for DcR have previously been elucidated and are attributed to DcR-mediated stimulation of the Akt, ERK (Secchiero et al, 2003) and NF-κB (Degli-Esposti et al, 1997) survival pathways, competitive binding of TRAIL to DcR1 inhibiting DR-induced DISC formation and DcR2 forming ligand-independent complexes with DR5 at the DISC to inhibit DR4 corecruitment and caspase activation (Ashkenazi, 2008).
Our work demonstrates that the combination of fenretinide and DR ligands, actinomycin D and TRAIL and etoposide used in combination with either TRAIL or FasL may be advantageous for the treatment of ESFT cells. As DR ligands transduce apoptotic signals through different pathways to those of chemotherapy and irradiation, these combinations may augment patient response to either drug alone and so be more effective. The combination of fenretinide (Garaventa et al, 2003; Villablanca et al, 2006; Formelli et al, 2008) and TRAIL (Ashkenazi, 2008) is an attractive therapeutic strategy because both agents are well tolerated in clinical trials and non-malignant cells are resistant to their cytotoxic effects, unlike many commonly used agents that are used in combination with TRAIL. Furthermore, both agents induce cytotoxicity independently of p53 (Hail et al, 2006; Ashkenazi, 2008), which is important when p53 mutations are common in certain cancer types and many conventional treatments such as irradiation and DNA-damaging drugs rely on p53 to induce cell death.
The combination of FasL with fenretinide or etoposide may also have therapeutic potential. However, at present, this is not a viable combination in the clinic because FAS monoclonal antibodies have shown hepatotoxicity in preclinical trials (Ashkenazi, 2008). However, APO010 (a second generation FasL from TopoTarget) is a recombinant mega-FasL that displays anticancer activities both in vitro and in xenograft models of human cancer. A dose-escalation phase I trial is currently recruiting participants with solid tumours to examine APO010 toxicity (ClinicalTrials.gov Identifier: NCT00437736).
Two strategies are currently being explored for the clinical use of TRAIL: recombinant human preparations (Genentech, San Francisco, CA, USA) or monoclonal antibodies to DR4 (Mapatumumab, Human Genome Sciences, Rockville, MD, USA) and DR5 (Amgen, Daiichi Sankyo (Munich, Germany), Human Genome Sciences, Genentech and Novartis, Basel, Switzerland). Both show promise in clinical trials when used as monotherapies and in combination with agents such as chemotherapeutics, XIAP inhibitors, proteasome inhibitors, HDACi, natural products and BH3 mimetics (Ashkenazi, 2008). As mentioned previously, Mapatumumab only induced limited in vitro cytotoxicity and growth inhibition in ESFT xenograft tumour panels (Smith et al, 2009). Low DR4 mRNA expression was observed, in concordance with our results, suggesting that combination therapies of TRAIL receptor agonists with fenretinide or chemotherapeutic drugs would be a better therapeutic regime for the treatment of paediatric tumours because of the upregulation of DR or downregulation of antiapoptotic factors.
In summary, the synergistic cell death observed with the combined treatment of fenretinide with DR ligands is mediated through ASK1- and p38α-induced upregulation of DR at the cell surface. In contrast, only certain chemotherapeutic drugs upregulated DR at the cell surface, which did not always correspond to enhanced cell death when cells were cotreated with the cognate DR ligand. These data suggest that fenretinide in combination with TRAIL may be a particularly attractive therapeutic strategy, as both agents are well tolerated in clinical trials and both induce death through separate apoptotic pathways that may overcome chemoresistance.
Change history
29 March 2012
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Appierto V, Tiberio P, Villani MG, Cavadini E, Formelli F (2009) PLAB induction in fenretinide-induced apoptosis of ovarian cancer cells occurs via a ROS-dependent mechanism involving ER stress and JNK activation. Carcinogenesis 30: 824–831
Ashkenazi A (2008) Directing cancer cells to self-destruct with pro-apoptotic receptor agonists. Nat Rev Drug Discov 7: 1001–1012
Brownhill SC, Taylor C, Burchill SA (2007) Chromosome 9p21 gene copy number and prognostic significance of p16 in ESFT. Br J Cancer 96: 1914–1923
Chen JJ, Chou CW, Chang YF, Chen CC (2008) Proteasome inhibitors enhance TRAIL-induced apoptosis through the intronic regulation of DR5: involvement of NF-kappa B and reactive oxygen species-mediated p53 activation. J Immunol 180: 8030–8039
Coulthard LR, White DE, Jones DL, McDermott MF, Burchill SA (2009) p38(MAPK): stress responses from molecular mechanisms to therapeutics. Trends Mol Med 15: 369–379
Cuello M, Coats AO, Darko I, Ettenberg SA, Gardner GJ, Nau MM, Liu JR, Birrer MJ, Lipkowitz S (2004) N-(4-hydroxyphenyl) retinamide (4HPR) enhances TRAIL-mediated apoptosis through enhancement of a mitochondrial-dependent amplification loop in ovarian cancer cell lines. Cell Death Differ 11: 527–541
Das A, Banik NL, Ray SK (2008) N-(4-Hydroxyphenyl) retinamide induced both differentiation and apoptosis in human glioblastoma T98G and U87 MG cells. Brain Res 1227: 207–215
Debatin KM, Krammer PH (2004) Death receptors in chemotherapy and cancer. Oncogene 23: 2950–2966
Degli-Esposti MA, Dougall WC, Smolak PJ, Waugh JY, Smith CA, Goodwin RG (1997) The novel receptor TRAIL-R4 induces NF-kappaB and protects against TRAIL-mediated apoptosis, yet retains an incomplete death domain. Immunity 7: 813–820
Formelli F, Cavadini E, Luksch R, Garaventa A, Villani MG, Appierto V, Persiani S (2008) Pharmacokinetics of oral fenretinide in neuroblastoma patients: indications for optimal dose and dosing schedule also with respect to the active metabolite 4-oxo-fenretinide. Cancer Chemother Pharmacol 62: 655–665
Garaventa A, Luksch R, Lo Piccolo MS, Cavadini E, Montaldo PG, Pizzitola MR, Boni L, Ponzoni M, Decensi A, De Bernardi B, Bellani FF, Formelli F (2003) Phase I trial and pharmacokinetics of fenretinide in children with neuroblastoma. Clin Cancer Res 9: 2032–2039
Georgakis GV, Li Y, Humphreys R, Andreeff M, O’Brien S, Younes M, Carbone A, Albert V, Younes A (2005) Activity of selective fully human agonistic antibodies to the TRAIL death receptors TRAIL-R1 and TRAIL-R2 in primary and cultured lymphoma cells: induction of apoptosis and enhancement of doxorubicin- and bortezomib-induced cell death. Br J Haematol 130: 501–510
Gibson SB, Oyer R, Spalding AC, Anderson SM, Johnson GL (2000) Increased expression of death receptors 4 and 5 synergizes the apoptosis response to combined treatment with etoposide and TRAIL. Mol Cell Biol 20: 205–212
Hail Jr N, Kim HJ, Lotan R (2006) Mechanisms of fenretinide-induced apoptosis. Apoptosis 11: 1677–1694
Hemmings C (2010) The elaboration of a critical framework for understanding cancer: the cancer stem cell hypothesis. Pathology 42: 105–112
Hetschko H, Voss V, Seifert V, Prehn JH, Kogel D (2008) Upregulation of DR5 by proteasome inhibitors potently sensitizes glioma cells to TRAIL-induced apoptosis. FEBS J 275: 1925–1936
Kang MH, Wan Z, Kang YH, Sposto R, Reynolds CP (2008) Mechanism of synergy of N-(4-hydroxyphenyl)retinamide and ABT-737 in acute lymphoblastic leukemia cell lines: Mcl-1 inactivation. J Natl Cancer Inst 100: 580–595
Khwaja FS, Quann EJ, Pattabiraman N, Wynne S, Djakiew D (2008) Carprofen induction of p75NTR-dependent apoptosis via the p38 mitogen-activated protein kinase pathway in prostate cancer cells. Mol Cancer Ther 7: 3539–3545
Kim HJ, Chakravarti N, Oridate N, Choe C, Claret FX, Lotan R (2006) N-(4-hydroxyphenyl)retinamide-induced apoptosis triggered by reactive oxygen species is mediated by activation of MAPKs in head and neck squamous carcinoma cells. Oncogene 25: 2785–2794
Kontny U (2006) Regulation of apoptosis and proliferation in Ewing's sarcoma—opportunities for targeted therapy. Hematol Oncol 24: 14–21
Kouhara J, Yoshida T, Nakata S, Horinaka M, Wakada M, Ueda Y, Yamagishi H, Sakai T (2007) Fenretinide up-regulates DR5/TRAIL-R2 expression via the induction of the transcription factor CHOP and combined treatment with fenretinide and TRAIL induces synergistic apoptosis in colon cancer cell lines. Int J Oncol 30: 679–687
Kuma Y, Sabio G, Bain J, Shpiro N, Marquez R, Cuenda A (2005) BIRB796 inhibits all p38 MAPK isoforms in vitro and in vivo. Journal of Biological Chemistry 280: 19472–19479
Kwon D, Choi K, Choi C, Benveniste EN (2008) Hydrogen peroxide enhances TRAIL-induced cell death through up-regulation of DR5 in human astrocytic cells. Biochem Biophys Res Commun 372: 870–874
Lissat A, Vraetz T, Tsokos M, Klein R, Braun M, Koutelia N, Fisch P, Romero ME, Long L, Noellke P, Mackall CL, Niemeyer CM, Kontny U (2007) Interferon-gamma sensitizes resistant Ewing's sarcoma cells to tumor necrosis factor apoptosis-inducing ligand-induced apoptosis by up-regulation of caspase-8 without altering chemosensitivity. Am J Pathol 170: 1917–1930
Lu G, Punj V, Chaudhary PM (2008) Proteasome inhibitor Bortezomib induces cell cycle arrest and apoptosis in cell lines derived from Ewing's sarcoma family of tumors and synergizes with TRAIL. Cancer Biol Ther 7: 603–608
Maeda T, Hao C, Tron VA (2001) Ultraviolet light (UV) regulation of the TNF family decoy receptors DcR2 and DcR3 in human keratinocytes. J Cutan Med Surg 5: 294–298
Mendoza FJ, Ishdorj G, Hu X, Gibson SB (2008) Death receptor-4 (DR4) expression is regulated by transcription factor NF-kappaB in response to etoposide treatment. Apoptosis 13: 756–770
Merchant MS, Yang X, Melchionda F, Romero M, Klein R, Thiele CJ, Tsokos M, Kontny HU, Mackall CL (2004) Interferon gamma enhances the effectiveness of tumor necrosis factor-related apoptosis-inducing ligand receptor agonists in a xenograft model of Ewing's sarcoma. Cancer Res 64: 8349–8356
Myatt SS, Redfern CP, Burchill SA (2005) p38MAPK-Dependent sensitivity of Ewing's sarcoma family of tumors to fenretinide-induced cell death. Clin Cancer Res 11: 3136–3148
Nakamura T, Tanaka K, Matsunobu T, Okada T, Nakatani F, Sakimura R, Hanada M, Iwamoto Y (2007) The mechanism of cross-resistance to proteasome inhibitor bortezomib and overcoming resistance in Ewing's family tumor cells. Int J Oncol 31: 803–811
Ng CP, Zisman A, Bonavida B (2002) Synergy is achieved by complementation with Apo2L/TRAIL and actinomycin D in Apo2L/TRAIL-mediated apoptosis of prostate cancer cells: role of XIAP in resistance. Prostate 53: 286–299
Osone S, Hosoi H, Kuwahara Y, Matsumoto Y, Iehara T, Sugimoto T (2004) Fenretinide induces sustained-activation of JNK/p38 MAPK and apoptosis in a reactive oxygen species-dependent manner in neuroblastoma cells. Int J Cancer 112: 219–224
Picarda G, Lamoureux F, Geffroy L, Delepine P, Montier T, Laud K, Tirode F, Delattre O, Heymann D, Redini F (2010) Preclinical evidence that use of TRAIL in Ewing's sarcoma and osteosarcoma therapy inhibits tumor growth, prevents osteolysis, and increases animal survival. Clin Cancer Res 16: 2363–2374
Proctor A, Brownhill SC, Burchill SA (2009) The promise of telomere length, telomerase activity and its regulation in the translocation-dependent cancer ESFT; clinical challenges and utility. Biochim Biophys Acta 1792: 260–274
Quann EJ, Khwaja F, Djakiew D (2007) The p38 MAPK pathway mediates aryl propionic acid induced messenger rna stability of p75 NTR in prostate cancer cells. Cancer Res 67: 11402–11410
Secchiero P, Gonelli A, Carnevale E, Milani D, Pandolfi A, Zella D, Zauli G (2003) TRAIL promotes the survival and proliferation of primary human vascular endothelial cells by activating the Akt and ERK pathways. Circulation 107: 2250–2256
Shetty S, Gladden JB, Henson ES, Hu X, Villanueva J, Haney N, Gibson SB (2002) Tumor necrosis factor-related apoptosis inducing ligand (TRAIL) up-regulates death receptor 5 (DR5) mediated by NFkappaB activation in epithelial derived cell lines. Apoptosis 7: 413–420
Smith MA, Morton CL, Kolb EA, Gorlick R, Keir ST, Carol H, Lock R, Kang MH, Reynolds CP, Maris JM, Watkins AE, Houghton PJ (2009) Initial testing (stage 1) of mapatumumab (HGS-ETR1) by the pediatric preclinical testing program. Pediatr Blood Cancer 54: 307–310
Song JH, Kandasamy K, Kraft AS (2008) ABT-737 induces expression of the death receptor 5 and sensitizes human cancer cells to TRAIL-induced apoptosis. J Biol Chem 283: 25003–25013
Sonnemann J, Dreyer L, Hartwig M, Palani CD, Hong le TT, Klier U, Broker B, Volker U, Beck JF (2007) Histone deacetylase inhibitors induce cell death and enhance the apoptosis-inducing activity of TRAIL in Ewing′s sarcoma cells. J Cancer Res Clin Oncol 133: 847–858
Su RY, Chi KH, Huang DY, Tai MH, Lin WW (2008) 15-deoxy-Delta12, 14-prostaglandin J2 up-regulates death receptor 5 gene expression in HCT116 cells: involvement of reactive oxygen species and C/EBP homologous transcription factor gene transcription. Mol Cancer Ther 7: 3429–3440
Thomadaki H, Floros KV, Scorilas A (2009) Molecular response of HL-60 cells to mitotic inhibitors vincristine and taxol visualized with apoptosis-related gene expressions, including the new member BCL2L12. Ann N Y Acad Sci 1171: 276–283
Toscano F, Fajoui ZE, Gay F, Lalaoui N, Parmentier B, Chayvialle JA, Scoazec JY, Micheau O, Abello J, Saurin JC (2008) P53-mediated upregulation of DcR1 impairs oxaliplatin/TRAIL-induced synergistic anti-tumour potential in colon cancer cells. Oncogene 27: 4161–4171
Villablanca JG, Krailo MD, Ames MM, Reid JM, Reaman GH, Reynolds CP (2006) Phase I trial of oral fenretinide in children with high-risk solid tumors: a report from the Children's Oncology Group (CCG 09709). J Clin Oncol 24: 3423–3430
Westwood G, Dibling BC, Cuthbert-Heavens D, Burchill SA (2002) Basic fibroblast growth factor (bFGF)-induced cell death is mediated through a caspase-dependent and p53-independent cell death receptor pathway. Oncogene 21: 809–824
White DE, Burchill SA (2008) BAY 11-7082 induces cell death through NF-kappaB-independent mechanisms in the Ewing's sarcoma family of tumours. Cancer Lett 268: 212–224
Williamson AJ, Dibling BC, Boyne JR, Selby P, Burchill SA (2004) Basic fibroblast growth factor-induced cell death is effected through sustained activation of p38MAPK and up-regulation of the death receptor p75NTR. J Biol Chem 279: 47912–47928
Woo IS, Eun SY, Jang HS, Kang ES, Kim GH, Kim HJ, Lee JH, Chang KC, Kim JH, Han CW, Seo HG (2009) Identification of ADP-ribosylation factor 4 as a suppressor of N-(4-hydroxyphenyl)retinamide-induced cell death. Cancer Lett 276: 53–60
Woo SH, Park IC, Park MJ, An S, Lee HC, Jin HO, Park SA, Cho H, Lee SJ, Gwak HS, Hong YJ, Hong SI, Rhee CH (2004) Arsenic trioxide sensitizes CD95/Fas-induced apoptosis through ROS-mediated upregulation of CD95/Fas by NF-kappaB activation. Int J Cancer 112: 596–606
Yamaguchi H, Wang HG (2004) CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J Biol Chem 279: 45495–45502
Yodkeeree S, Sung B, Limtrakul P, Aggarwal BB (2009) Zerumbone enhances TRAIL-induced apoptosis through the induction of death receptors in human colon cancer cells: evidence for an essential role of reactive oxygen species. Cancer Res 69: 6581–6589
Yoshida S, Narita T, Koshida S, Ohta S, Takeuchi Y (2003) TRAIL/Apo2L ligands induce apoptosis in malignant rhabdoid tumor cell lines. Pediatr Res 54: 709–717
Acknowledgements
We thank Thomas Maisey and Andrea Berry for routine maintenance, characterisation and mycoplasm testing of cell lines, Dr James Boyne for the fenretinide array data, Drs Elena Jones, Eva Pitt, Ewan Morrison and Poulam Patel for the kind gift of cell lines and Mr Colin Johnston for statistical advice.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary information accompanies the paper on British Journal of Cancer website
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
White, D., Burchill, S. Fenretinide-dependent upregulation of death receptors through ASK1 and p38α enhances death receptor ligand-induced cell death in Ewing's sarcoma family of tumours. Br J Cancer 103, 1380–1390 (2010). https://doi.org/10.1038/sj.bjc.6605896
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6605896
Keywords
This article is cited by
-
CD271 activation prevents low to high-risk progression of cutaneous squamous cell carcinoma and improves therapy outcomes
Journal of Experimental & Clinical Cancer Research (2023)
-
Anti-cancer Effect of Hyoscyamus muticus Extract via Its Activation of Fas/FasL-ASK1-p38 Pathway
Biotechnology and Bioprocess Engineering (2022)
-
Targeting incretin hormones and the ASK-1 pathway as therapeutic options in the treatment of non-alcoholic steatohepatitis
Hepatology International (2018)
-
The alkyllysophospholipid edelfosine enhances TRAIL-mediated apoptosis in gastric cancer cells through death receptor 5 and the mitochondrial pathway
Tumor Biology (2016)
-
Fenretinide sensitizes multidrug-resistant human neuroblastoma cells to antibody-independent and ch14.18-mediated NK cell cytotoxicity
Journal of Molecular Medicine (2013)
