
Abstract
Background:
We evaluated bevacizumab with metronomic etoposide among recurrent malignant glioma patients in a phase 2, open-label trial.
Methods:
A total of59 patients, including 27 with glioblastoma (GBM) and 32 with grade 3 malignant glioma, received 10 mg kg−1 bevacizumab biweekly and 50 mg m−2 etoposide daily for 21 consecutive days each month. The primary end point was a 6-month progression-free survival, and secondary end points included safety and overall survival. Vascular endothelial growth factor (VEGF), VEGFR-2, carbonic anhydrase 9 (CA9) and hypoxia-inducible factor-2α (HIF-2α) were assessed semiquantitatively in archival tumours using immunohistochemistry and were correlated with outcome.
Results:
Among grade 3 and GBM patients, the 6-month progression-free survivals were 40.6% and 44.4%, the radiographic response rates were 22% and 37% and the median survivals were 63.1 and 44.4 weeks, respectively. Hypertension predicted better outcome among both grade 3 and GBM patients, whereas high CA9 and low VEGF were associated with poorer progression-free survival (PFS) among those with GBM. The most common grade ⩾3 adverse events included neutropaenia (24%), thrombosis (12%), infection (8%) and hypertension (3%). Two patients had asymptomatic, grade 1 intracranial haemorrhage and one on-study death occurred because of pulmonary embolism.
Conclusion:
Bevacizumab with metronomic etoposide has increased toxicity compared with previous reports of bevacizumab monotherapy. Its anti-tumour activity is similar to that of bevacizumab monotherapy or bevacizumab plus irinotecan. (ClinicalTrials.gov: NCT00612430).
Similar content being viewed by others

Main
Malignant gliomas, including the most common subtype, grade 4 glioblastoma (GBM), as well as WHO grade 3 tumours such as anaplastic astrocytoma (AA), remain essentially fatal malignancies (Wen and Kesari, 2008). The median survival for GBM patients is <15 months after external beam radiotherapy and temozolomide after maximal safe resection (Stupp et al, 2005), whereas survival for most grade 3 patients is 2–3 years (See and Gilbert, 2004).
Profound angiogenesis, a hallmark of malignant glioma, results from a ‘perfect storm’ of hypoxia and hypoxia-independent aberrant activation of cell signalling pathways that markedly upregulate vascular endothelial growth factor (VEGF) expression (Jain et al, 2007). Anti-angiogenic strategies targeting VEGF are active among recurrent GBM patients. Daily administration of AZD2171 (cediranib, AstraZeneca Pharmaceuticals, London, UK), a potent oral, pan-VEGF receptor inhibitor that also inhibits platelet-derived growth factor (PDGF) receptor and c-Kit, achieved an overall radiographic response (ORR) of 56% and a median progression-free survival (PFS) of 3.96 months (Batchelor et al, 2007). Bevacizumab, a humanised monoclonal antibody against VEGF (Avastin, Genentech Pharmaceuticals, South San Francisco, CA, USA), was originally reported to achieve dramatic radiographic responses when combined with the topoisomerase-1 inhibitor irinotecan among recurrent GBM patients (Stark-Vance, 2005). In a formal phase II study of this regimen, Vredenburgh noted a 61% ORR, 46% 6-month PFS (6-PFS) and a median overall survival (OS) of 10 months among heavily pre-treated, recurrent GBM patients (Vredenburgh et al, 2007a, 2007b). The anti-tumour activity of bevacizumab with and without irinotecan was confirmed in two follow-up, prospective studies (Friedman et al, 2009; Kreisl et al, 2009) that recently led to accelerated approval of bevacizumab monotherapy for recurrent GBM by the US Food and Drug Administration. In addition, we have also recently noted that bevacizumab plus irinotecan is active for recurrent grade 3 malignant glioma patients (Desjardins et al, 2008). These studies, as well as additional retrospective series (Bokstein et al, 2008; Kang et al, 2008; Zuniga et al, 2009), have also confirmed the safety profile of this regimen for malignant glioma patients.
This study was conducted to evaluate the anti-tumour activity of etoposide, a topoisomerase-2 inhibitor, when administered on a protracted, oral, ‘metronomic’ dosing schedule, in combination with standard bevacizumab administration. Etoposide has modest activity among recurrent malignant glioma patients as either single-agent therapy or in combination with other chemotherapeutics (Chamberlain and Grafe, 1995; Fulton et al, 1996; Kesari et al, 2007). We hypothesised that etoposide administered on protracted, daily schedule may exert an effect as a metronomic chemotherapeutic (Kerbel and Kamen, 2004) and thereby enhance the anti-angiogenic and anti-tumour effect of bevacizumab in this patient population.
Patients and methods
Protocol objectives
Our primary objective was to estimate the 6-month PFS among adults with recurrent malignant glioma treated with protracted, oral etoposide plus bevacizumab. Secondary objectives included evaluation of the safety and tolerability of this regimen and evaluation of the radiographic response, PFS and OS associated with this regimen in this patient population.
Patient eligibility
Patients were required to have histological confirmation of grade 3 or 4 malignant glioma that was recurrent after previous radiation or chemotherapy. Patients with previous low-grade glioma were eligible if histological transformation to malignant glioma was confirmed. Eligible patients were also at least 18 years of age, had a Karnofsky performance status (KPS) ⩾60% and were on a stable corticosteroid dose for at least 1 week. Additional enrollment criteria included: haematocrit >29%; absolute neutrophil count >1500 cells μl−1; platelet count >100 000 cells μl−1; and serum creatinine <1.5 mg per 100 ml, aspartate aminotransferase and bilirubin <1.5 times the institutional upper limit of normal. At least 4 weeks between surgical resection or chemotherapy, and at least 12 weeks between radiotherapy, and enrollment were required. All patients provided informed consent.
Patients were excluded for: >3 recurrences; uncontrolled hypertension; therapeutic anticoagulation use; acute haemorrhage on baseline MRI; urine protein:creatinine ratio >1; pregnancy or nursing; previous bevacizumab or etoposide; active infection requiring intravenous antibiotics; and previous stereotactic radiosurgery, radiation implants or radiolabelled monoclonal antibody therapy, unless there was unequivocal disease progression (such as a new lesion or biopsy-proven recurrence).
Treatment design
Patients were stratified by histopathology into those with GBM and those with grade 3 malignant glioma. All patients were administered 50 mg m−2 of etoposide daily for 21 consecutive days of each 28-day cycle. Bevacizumab was administered at 10 mg kg−1 intravenously every 14 days. Study therapy was planned to discontinue after 12 cycles. However, patients were given the option to continue study therapy for 6 additional months if they were felt to potentially derive further therapeutic benefit. Specifically, such patients included those who had ongoing radiographic or clinical improvement after 12 months or those who had persistent contrast enhancement that was accompanied by hypermetabolic activity on [18F]fluorodeoxyglucose positron emission tomography (18FDG-PET) imaging. Study therapy was discontinued for unacceptable toxicity, tumour progression, study non-compliance or consent withdrawal.
Response evaluation
Study investigators determined response by neurological examination and contrast-enhanced MRI previous to every other treatment cycle. A complete response (CR) required disappearance of all enhancing tumours with corticosteroid discontinuation and neurological stability or improvement. A partial response (PR) required ⩾50% reduction in size (product of largest perpendicular diameters) of enhancing tumour with stability or improvement of neurological status and corticosteroids. Complete and partial responses also required stable or improved signal abnormality on fluid-attenuated inversion recovery (FLAIR) sequences and confirmation on consecutive scans at least 4 weeks apart. Progressive disease (PD) was defined by any of the following: ⩾25% increase on enhancing tumour; a new enhancing lesion; or clear worsening of FLAIR signal abnormality or significant clinical decline that was not attributable to co-morbid event or concurrent medication. Stable disease was defined as any assessment not meeting CR, PR or PD criteria. The inclusion of FLAIR signal changes in the classification of response in this study was based on the growing recognition that some malignant glioma patients undergoing therapy with VEGF or VEGFR inhibitors may have significant improvement in contrast enhancement, yet have extensive progressive changes on T2 or FLAIR sequences, often accompanied by marked neurological decline, which is felt to be indicative of progressive infiltrative tumour (Bokstein et al, 2008; Norden et al, 2008; van den Bent et al, 2009).
Dose modification and retreatment criteria
Daily etoposide dose was reduced by 25 mg for grade 3 thrombocytopenia, grade 4 neutropaenia or other related grade ⩾3 non-haematologic events. Bevacizumab was discontinued for uncontrollable hypertension, grade 2 or greater haemorrhage, arterial thrombosis, wound dehiscence requiring surgical intervention, intestinal perforation, grade 4 venous thrombosis, proteinuria or congestive heart failure. Bevacizumab was held until other grade 3, related events resolved to a grade ⩽ 1.
Initiation of each cycle required an absolute neutrophil count (ANC) ⩾1000 mm−3; a platelet count ⩾100 000 mm−3; aspartate aminotransferase (AST), bilirubin and creatinine less than twice the institutional upper limit of normal; and resolution of any related grade ⩾3 event to a grade ⩽1.
Tumour marker analysis
Immunohistochemistry staining of archival, paraffin-embedded tumour sections was performed and analysed for VEGF, VEGFR-2/KDR as well as two markers of hypoxia, hypoxia-inducible factor 2α (HIF-2α) and carbonic anhydrase 9 (CA9), as previously described (Sathornsumetee et al, 2008) by a neuropathologist who was unaware of clinical and imaging outcome. A semiquantitative score was derived from an intensity score of the reactivity product (absent, 0; mild, 1; moderate, 2; strong, 3) related with endogenous positive controls multiplied by distribution score (percentage of reactive cells in tumour).
Statistical considerations
The primary aim of this study was to evaluate the 6-PFS rate of bevacizumab plus protracted, oral etoposide among recurrent malignant glioma patients. Yung et al (2000) reported a 6-PFS rate of 21% (95% CI 13–29) among recurrent GBM patients treated with temozolomide at first recurrence. Given that the prognosis of our patient population was expected to be poorer than that reported by Yung et al, the sample size goal of 27 GBM patients for this study was chosen to provide 82% power to differentiate between 6-PFS of 5 and 20% with a type I error of 0.04. Similarly, the 6-PFS of temozolomide for recurrent grade 3 malignant glioma patients as reported by Yung et al is 46% (95% CI 38–54). Given that the prognosis of our grade 3 patients was again expected to be poorer than that reported, a sample size goal of 32 recurrent grade 3 patients was chosen to provide 80% power to differentiate between a 6-PFS of 20 and 40% with a type I error rate of 0.04. The benchmark set by temozolomide was chosen as the historical comparator for our study rather than the outcome reported on previous bevacizumab studies because the latter had not been validated in a multi-institutional setting when this study was designed.
‘Stopping rules’ for poor efficacy and unacceptable toxicity were incorporated for each stratum. Specifically, if ⩾10 of the first 16 patients per stratum progressed or died within 2 months of study initiation, further accrual would be suspended. In addition, if 6 or more of the first 16 patients per stratum experienced unacceptable toxicity, defined as grade ⩾4 non-haematological events, further accrual would be suspended.
Progression-free survival and OS were measured from the cycle 1 start date and summarised using Kaplan–Meier estimator including 95% CIs. For each cohort, PFS distribution was compared between the following subgroups using the log-rank test: patients <50 years old vs those ⩾50 years; patients with a KPS <90 vs those with a KPS ⩾90; patients with >1 previous episode of progression vs those with 1 previous progression; and patients who received >1 previous chemotherapeutic vs those who received only 1 previous chemotherapeutic. We also sought to determine whether hypertension was associated with outcome. For these purposes, hypertension was defined as sustained grade 1 for at least 4 weeks, grade ⩾2 or the initiation or increase in anti-hypertensive medications. Log-rank tests were conducted comparing patients who developed hypertension with those who did not relative to OS and PFS.
The effect of each tumour marker on overall and PFS was evaluated using separate Cox's proportional hazard models. Hazard ratios and the P-value from Wald's chi-square tests were determined for each model. The effect of each tumour marker (CA9, VEGF, VEGFR-2 and HIF2α) on PFS-6 and radiographic response was evaluated using logistic regression models. Odds ratios and the P-value from Wald's chi-square tests were determined for each model. The effect of each categorical marker on PFS-6 and radiographic response was also estimated using Fisher's exact test.
Results
Patient characteristics
A total of 59 patients were enrolled between April 2007 and January 2008, including 32 with grade 3 malignant glioma and 27 with GBM (Table 1).
Most patients had bulky disease at enrollment, given that only 7% underwent a gross total resection of enhancing disease before enrollment. Patients who underwent a GTR were not evaluable for radiographic response. In general, patients were significantly pretreated with 53% enrolling at either second or third progression, whereas 73% had received two or more previous chemotherapeutic agents.
As of 15 May 2009, four GBM patients (15%) remain alive, including three with progressive disease and one with stable disease in the tenth month of the study therapy. Again, 16 grade 3 malignant glioma patients (50%) remain alive, including 2 who continue on-study therapy, 5 who are off study without recurrence and 9 with progressive disease. In total, 39 patients (66%) have died. Median follow-up was 91.2 weeks for GBM patients, and 69.9 weeks for grade 3 malignant glioma patients.
Study drug administration and safety
Study drug administration and compliance with treatment for the intent-to-treat (ITT) study population were excellent. A total of 389 cycles of therapy were administered, including a median of 6.6 (range 1–18) cycles for the grade 3 malignant glioma patients and 6.6 (range 1–20) cycles for the GBM patients, respectively.
All patients were assessable for toxicity. As the adverse event profiles were similar for both patient cohorts, a pooled summary of toxicity is provided in Table 2. Significant haematological toxicity included neutropaenia (grade 3, n=9 (15%); grade 4, n=5 (8%)), thrombocytopaenia (grade 3, n=2 (3%)) and anaemia (grade 3, n=1 (2%)). Most non-haematologic toxicities were of grade 2. The most common serious non-haematologic events were thrombosis (grade 3, n=4 (7%); grade 4, n=2 (3%); grade 5, n=1 (2%)), infection (grade 3, n=4 (7%); grade 4, n=1 (2%)) and hypertension (grade 3, n=1 (2%); grade 4, n=1 (2%)). Haemorrhage was limited to two patients with grade 1 central nervous system (CNS) bleeds detected on surveillance MRI, and one patient with grade 2 GI bleeding from haemorrhoids. A single on-study death, attributed to pulmonary embolism, occurred.
A total of nine grade 3 malignant glioma patients (28%) required etoposide dose modification, including eight patients for grade 3 or 4 neutropaenia and one patient with grade 2 peripheral neuropathy. Two GBM patients (7%) required etoposide dose modification including one for grade 4 neutropaenia and one for peripheral neuropathy. Four patients (13%) on the grade 3 arm discontinued therapy because of toxicity including one patient with persistent grade 3 proteinuria after 14 cycles of therapy, 1 patient with dehiscence of a previously healed craniotomy incision after 7 cycles of therapy and the two patients described above with grade 1 CNS haemorrhage. Three GBM patients (11%) discontinued therapy because of adverse events, all of whom had pulmonary emboli.
Outcome
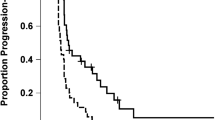
The median OS, PFS, 6-PFS rate and the rate of radiographic response are summarised in Table 3 and Figure 1. Outcome comparisons were not performed for subsets of grade 3 malignant glioma patients because of the small sample size. Complete and partial responses were observed in one (4%) and five (19%) GBM patients, and two (7%) and five (17%) grade 3 patients. Stable disease was the best response in 19 (73%) and 21 (72%) of GBM and grade 3 patients, respectively, whereas only two GBM (7%) and four grade 3 (13%) patients progressed at first evaluation. In total, 10 patients (17%) completed 12 cycles of therapy and eight of these patients (80%) had no evidence of hypermetabolic activity on [18F]FDG-PET imaging (Figure 2A). Among six patients (10%) who completed 1 year of planned therapy, five remain alive, including four without tumour recurrence (all grade 3 malignant glioma) and one (GBM) with recurrent tumour; one GBM patient who completed a year of therapy subsequently died of progressive tumour.

Kaplan–Meier plots of progression-free survival (A) and overall survival (B) for grade 3 malignant glioma and glioblastoma patients.

Baseline and post-treatment magnetic resonance imaging of a representative, responding glioblastoma patient including post-contrast axial T1-weighted and fluid-attenuated inversion recovery (FLAIR) images. [18F]fluorodeoxyglucose positron-emission tomography imaging at the completion of 12 cycles of therapy shows no evidence of hypermetabolic activity (arrow). (A) Baseline and off-study magnetic resonance imaging showing worsened FLAIR signal abnormality, indicating an infiltrative, microscopic pattern of treatment failure (arrows) involving the isolateral hemisphere (B) and the contralateral hemisphere (C), despite improved contrast enhancement.
Although limited by sample size, the development of grade ⩾1 hypertension was linked with improved outcome. Among GBM patients who developed hypertension, median OS was not reached and 1-year OS was 100%, whereas median and 1-year OS were 39.4 weeks and 34.8%, respectively, for those who did not develop hypertension (P=0.0095). A trend towards improved OS was also noted among grade 3 patients who developed hypertension and all of these patients also remain alive. In addition, PFS was longer for grade 3 patients who developed hyptertension (P=0.024). Among GBM patients, median and 6-PFS were 61 weeks and 100%, respectively, for those who developed hypertension when compared with 16 weeks and 34.8% for those who did not; however, these differences did not achieve statistical significance (P=0.123).
None of the demographic or pre-treatment characteristics had an association with PFS within either patient cohort, although a trend towards improved PFS was noted among grade 3 patients with a KPS ⩾90 (P=0.054). In all, eight patients developed hypertension (four GBM and four grade 3 MG, Table 2). A trend towards improved survival was observed in these patients, but the sample size was too small for subgroup analysis.
The 27 patients who achieved stable disease or radiographic response were on corticosteroids at study initiation, and 10of them (37%) decreased corticosteroids while on study, including five patients (19%) who successfully tapered off corticosteroids completely.
Tumour markers
In all, 12 GBM patients (44%) and 11 grade 3 malignant glioma patients (34%) had adequate tumour material for marker expression analysis (Supplementary Table 1, Supplementary Figure 1). All tumours expressed VEGF and VEGFR2 but the range of expression varied from 10 to 80% for VEGF and 2 to 80% for VEGFR2. Carbonic anhydrase 9 was detected in 11 of 12 (92%) GBM tumours (percentage of positive cells: 1–80%) and in 8 of 11 (73%) grade 3 tumours (range of positive cells: 1–40%). Hypoxia-inducible factor-2α was detected in 8 of 8 GBM tumours (range of positive cells: 0.01–4%) and in 8 of 10 grade 3 tumours (range of positive cells: 0.1–20%). All markers were more commonly expressed by GBM tumours compared with grade 3 tumours.
Low CA9 expression (⩽10% of cells; P=0.04) and increased VEGF expression (>30% of cells; P=0.006) were associated with better PFS among GBM patients (Supplementary Table 1). None of the tumour markers were predictive of outcome among grade 3 tumours.
Pattern of failure
A total of 22 grade 3 malignant glioma patients (69%) and 21 GBM patients (78%) developed radiographic evidence of progressive disease. The pattern of recurrence included worsened enhancement plus FLAIR changes at the primary tumour site in 15 (68%) of the grade 3 patients and 13 (62%) GBM patients. Clinical decline and progressive local FLAIR changes without worsened enhancement were noted in four grade 3 patients (18%) and six GBM patients (29%), respectively (Figures 2B and C). In addition, three grade 3 patients (14%) and two GBM patients (10%) had new distant sites of disease (defined as >2 cm away from previous enhancement without contiguous FLAIR signal abnormality) at progression.
Discussion
We report the first clinical trial evaluating metronomic chemotherapy combined with bevacizumab for recurrent malignant glioma patients. We hypothesised that etoposide, administered on a metronomic or protracted, daily dosing schedule would enhance the anti-tumour activity of bevacizumab based on potentially complimentary mechanisms of anti-angiogenic action. Bevacizumab binds VEGF-A, preventing activation of VEGFR1 and VEGFR2, whereas metronomic chemotherapy may exert an effect through a number of anti-angiogenic actions, including induction of endogenous angiogenesis inhibitors such as thrombospondin-1 (Damber et al, 2006), enhancement of endothelial cells apoptosis (Bocci et al, 2002) and decreasing the mobilisation and viability of circulating endothelial progenitor cells (Bertolini et al, 2003). Preclinical studies confirm that metronomic etoposide is preferentially cytotoxic against tumour endothelial cells compared with tumour cells in vitro (Drevs et al, 2004), and that metronomic etoposide plus anti-angiogenic therapy prolongs survival in orthotopic, intracranial U87 GBM xenografts compared with conventionally dosed chemotherapy with or without anti-angiogenic therapy (Bello et al, 2001). Clinically, several studies using metronomic dosing of etoposide have shown evidence of modest activity among recurrent malignant glioma patients (Chamberlain and Grafe, 1995; Fulton et al, 1996; Kesari et al, 2007), as well as other cancer patient populations (Correale et al, 2006; Twardowski et al, 2008). To date, the only published studies evaluating metronomic chemotherapy plus bevacizumab have involved patients with recurrent breast and ovarian cancer, and show anticancer benefit (Dellapasqua et al, 2008; Garcia et al, 2008; Garcia-Saenz et al, 2008).
Our study revealed that metronomic etoposide plus bevacizumab has encouraging outcome when compared with established benchmarks. Specifically, for recurrent GBM patients, our 6-PFS rate and median OS were higher than those reported with temozolomide at first recurrence (Yung et al, 2000), as well as several studies with etoposide (Fulton et al, 1996; Chamberlain and Kormanik, 1999) and historical series of salvage regimens.(Wong et al, 1999; Ballman et al, 2007; Lamborn et al, 2008). In addition, the outcomes of our study did not differ significantly to that achieved with bevacizumab plus irinotecan in a single-institution, phase 2 study (Vredenburgh et al, 2007b). It is noteworthy that patients in that study as well as in this study were heavily pretreated with a median of two previous episodes of progressive disease.
A major remaining question is whether chemotherapy, including metronomic, adds benefit over bevacizumab alone for recurrent malignant glioma patients. Although a recently reported phase II study randomised GBM patients at either first or second recurrence to bevacizumab monotherapy or bevacizumab plus irinotecan, this study was not powered to compare treatment arms and randomisation served solely to eliminate treatment assignment bias. The rate of radiographic response by blinded centralised review and 6-PFS were 38 and 50% for patients treated with combination therapy and 28 and 43% for those treated with bevacizumab monotherapy. Although median OS did not differ between the arms, monotherapy patients were also allowed to crossover to the combination arm at progression and continue study therapy. Of concern, the overall toxicity appeared increased in the combination cohort (Friedman et al, 2009). A recent single-arm study confirmed the anti-tumour efficacy of bevacizumab monotherapy among recurrent GBM patients (Kreisl et al, 2009). The results of the current study are comparable with those reported previously for bevacizumab-based therapy (Table 3). Furthermore, the addition of chemotherapy to bevacizumab, as shown in previous studies with irinotecan and in this study with metronomic etoposide, can lead to increased toxicity.
In this study we also noted that bevacizumab plus metronomic etoposide is active among recurrent grade 3 malignant glioma patients. It is noteworthy that there are no data with bevacizumab monotherapy for this subset of malignant glioma patients, but our study results compare favourably with those achieved with bevacizumab plus irinotecan (Desjardins et al, 2008) (Table 3), and are superior to those reported in two meta-analyses of salvage regimens (Wong et al, 1999; Lamborn et al, 2008).
As noted in other clinical trials (Chamberlain and Grafe, 1995; Fulton et al, 1996; Kesari et al, 2007), protracted daily etoposide was well tolerated and did not increase bevacizumab-specific toxicity. Grade 3 or 4 neutropaenia, the most common serious toxicity, occurred in approximately 24% of patients, but responded to dose modification. Grade 3 or 4 anaemia and thrombocytopaenia occurred rarely. The spectrum of non-haematological toxicity was comparable with that previously reported for bevacizumab in this patient population (Friedman et al, 2009) and appeared more favourable than that reported for bevacizumab plus irinotecan. Specifically, 7 patients (11%) in the current study discontinued therapy because of toxicity, whereas 11 patients (31%) in our single-institutional study (Vredenburgh et al, 2007b) and 14 patients (18%) in the randomised study (Friedman et al, 2009) discontinued bevacizumab plus irinotecan because of toxicity.
Corticosteroids, which are routinely administered to brain tumour patients to control symptoms due to cerebral oedema, cause a multitude of adverse sequellae that diminish quality of life, including marked weight gain, muscle loss, osteoporosis, hypertension, dysregulated glucose metabolism and immunosuppression (Wen et al, 2006). In this study, 37% of patients who were on chronic corticosteroid administration at the time of enrollment were able to taper their daily corticosteroid dose, including 19% who were able to completely discontinue corticosteroids. A similar ability to decrease chronic corticosteroid requirements has been noted in other studies evaluating anti-VEGF/R therapeutics (Batchelor et al, 2007; Vredenburgh et al, 2007a, 2007b; Desjardins et al, 2008).
Hypertension, a known complication of VEGFR signalling inhibition, is likely related with several factors, including diminished nitric oxide and prostacyclin synthesis (Scotland et al, 2005), diminished endothelial baroreceptor response (Yang et al, 2002), small artery and arteriole rarefaction (Ciuffetti et al, 2003) as well as increased vascular stiffness (Veronese et al, 2006). To our knowledge, this is the first clinical trial among malignant glioma patients to note an association between hypertension after VEGF-targeted therapy and improved outcome. However, this association should be interpreted cautiously and may have been because of a statistical coincidence, given the small number of patients who developed hypertension on this study. Clearly, our results require further validation in subsequent clinical trials. Nonetheless, hypertension has been associated with better outcome among other cancer patient populations after bevacizumab (Rixe et al, 2007; Schneider et al, 2008; Scartozzi et al, 2009), leading some to postulate that hypertension may serve as a biomarker of response (Jubb et al, 2006; van Heeckeren et al, 2007). Alternatively, it is possible that the development of hypertension may simply reflect longer bevacizumab exposure. We evaluated this possibility and noted that the interval from study initiation to hypertension was 6.4 weeks for GBM patients and 32 weeks for the grade 3 patients. The fact that both of these values were considerably less than the median PFS observed for each study cohort suggests that hypertension may be a predictive marker rather than simply reflecting prolonged bevacizumab exposure. We further analysed this association by reviewing our previous bevacizumab plus irinotecan trial for recurrent GBM (Vredenburgh et al, 2007b). In this series, similar to the current study, there was a trend towards improved median PFS, 6-PFS and OS among patients who developed hypertension compared with those who did not, but this analysis was also limited by small sample size. Nonetheless, further evaluation of hypertension as a potential predictor of bevacizumab activity among recurrent malignant glioma patients should be considered.
Our evaluation of tumour marker expression and outcome were clearly limited by the small number of analysed tumours as well as the use of archival tumour samples. Nonetheless, we noted that improved PFS was associated with lower CA9 expression as a marker of tumour hypoxia. It is noteworthy that we previously noted a similar association among recurrent malignant glioma patients treated with bevacizumab and irinotecan (Sathornsumetee et al, 2008). Of interest, we also noted that patients who had lower levels of VEGF, the target of bevacizumab, VEGF, seemed to respond less well with a lower PFS. Further analysis to validate these findings should be considered.
Our group and others have noted that potent VEGF inhibitors can decrease the permeability and intravenous contrast enhancement within 24–48 h (Batchelor et al, 2007; Desjardins et al, 2007). Although radiographic response among malignant glioma patients is traditionally based on changes in the largest bi-dimensional product of enhancement after gadolinium administration (Macdonald et al, 1990), anti-VEGF therapy can significantly and progressively worsen non-enhancing oedema on MRI that may be due to infiltrative disease, despite stable or even improved contrast-enhanced images (Ananthnarayan et al, 2008; Kang et al, 2008; Norden et al, 2008; Zuniga et al, 2009). We observed this pattern of progression in 17% of patients in the current study. Selection for a more invasive phenotype is supported by preclinical studies that revealed increased ‘satellites’ of infiltrative tumour that co-opt normal host vessels in orthotopic GBM xenografts treated with a VEGF monoclonal antibody (Rubenstein et al, 2000). Furthermore, as noted by others (Norden et al, 2008; Zuniga et al, 2009), we observed that new distant disease occurs in approximately 10% of patients. More diffuse, infiltrative, non-enhancing tumour at progression as well as the emergence of new, distant CNS sites of disease suggest that malignant gliomas may evolve resistance to VEGF-targeted therapeutics by adopting a more invasive phenotype that is relatively independent of VEGF signalling.
We performed the first study of bevacizumab administered with a metronomic chemotherapeutic (etoposide) and show that this regimen is safe and associated with encouraging clinical and survival benefit among recurrent GBM and grade 3 malignant glioma patients. Further studies evaluating this specific regimen as well as others, combining a metronomic chemotherapeutic plus a VEGF/R inhibitor, should be considered. Additional prospective studies to evaluate potential predictive biomarkers of anti-VEGF therapy for malignant glioma patients, including hypertension as well as tumour hypoxia and VEGF expression, are appropriate. Finally increased understanding of mechanisms of VEGF inhibitor resistance, including enhanced invasion and infiltration, is critically needed to further improve the outcome with anti-angiogenic therapy for malignant glioma patients.
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Ananthnarayan S, Bahng J, Roring J, Nghiemphu P, Lai A, Cloughesy T, Pope WB (2008) Time course of imaging changes of GBM during extended bevacizumab treatment. J Neurooncol 88: 339–347
Ballman KV, Buckner JC, Brown PD, Giannini C, Flynn PJ, LaPlant BR, Jaeckle KA (2007) The relationship between six-month progression-free survival and 12-month overall survival end points for phase II trials in patients with glioblastoma multiforme. Neuro Oncol 9: 29–38
Batchelor TT, Sorensen AG, di Tomaso E, Zhang WT, Duda DG, Cohen KS, Kozak KR, Cahill DP, Chen PJ, Zhu M, Ancukiewicz M, Mrugala MM, Plotkin S, Drappatz J, Louis DN, Ivy P, Scadden DT, Benner T, Loeffler JS, Wen PY, Jain RK (2007) AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell 11: 83–95
Bello L, Carrabba G, Giussani C, Lucini V, Cerutti F, Scaglione F, Landre J, Pluderi M, Tomei G, Villani R, Carroll RS, Black PM, Bikfalvi A (2001) Low-dose chemotherapy combined with an antiangiogenic drug reduces human glioma growth in vivo. Cancer Res 61: 7501–7506
Bertolini F, Paul S, Mancuso P, Monestiroli S, Gobbi A, Shaked Y, Kerbel RS (2003) Maximum tolerable dose and low-dose metronomic chemotherapy have opposite effects on the mobilization and viability of circulating endothelial progenitor cells. Cancer Res 63: 4342–4346
Bocci G, Nicolaou KC, Kerbel RS (2002) Protracted low-dose effects on human endothelial cell proliferation and survival in vitro reveal a selective antiangiogenic window for various chemotherapeutic drugs. Cancer Res 62: 6938–6943
Bokstein F, Shpigel S, Blumenthal DT (2008) Treatment with bevacizumab and irinotecan for recurrent high-grade glial tumors. Cancer 112: 2267–2273
Chamberlain MC, Grafe MR (1995) Recurrent chiasmatic-hypothalamic glioma treated with oral etoposide. J Clin Oncol 13: 2072–2076
Chamberlain MC, Kormanik PA (1999) Salvage chemotherapy with tamoxifen for recurrent anaplastic astrocytomas. Arch Neurol 56: 703–708
Ciuffetti G, Schillaci G, Innocente S, Lombardini R, Pasqualini L, Notaristefano S, Mannarino E (2003) Capillary rarefaction and abnormal cardiovascular reactivity in hypertension. J Hypertens 21: 2297–2303
Correale P, Cerretani D, Remondo C, Martellucci I, Marsili S, La Placa M, Sciandivasci A, Paolelli L, Pascucci A, Rossi M, Di Bisceglie M, Giorgi G, Gotti G, Francini G (2006) A novel metronomic chemotherapy regimen of weekly platinum and daily oral etoposide in high-risk non-small cell lung cancer patients. Oncol Rep 16: 133–140
Damber JE, Vallbo C, Albertsson P, Lennernas B, Norrby K (2006) The anti-tumour effect of low-dose continuous chemotherapy may partly be mediated by thrombospondin. Cancer Chemother Pharmacol 58: 354–360
Dellapasqua S, Bertolini F, Bagnardi V, Campagnoli E, Scarano E, Torrisi R, Shaked Y, Mancuso P, Goldhirsch A, Rocca A, Pietri E, Colleoni M (2008) Metronomic cyclophosphamide and capecitabine combined with bevacizumab in advanced breast cancer. J Clin Oncol 26: 4899–4905
Desjardins A, Barboriak DP, Herndon IJ, Reardon D, Quinn J, Rich J, Sathornsumetee S, Gururangan S, Friedman HS, Vredenburgh J (2007) Dynamic contrast-enhancement magnetic resonance imaging (DEC-MRI) evaluation in glioblastoma (GBM) patients treated with bevacizumab (BEV) and irinotecan (CPT-11). In ASCO Annual Meeting 2007. Proceedings of the American Society of Clinical Oncology, Grunberg SM (ed) Vol. 25, pp 82s. American Society of Clinical Oncology: Alexandria, VA
Desjardins A, Reardon DA, Herndon 2nd JE, Marcello J, Quinn JA, Rich JN, Sathornsumetee S, Gururangan S, Sampson J, Bailey L, Bigner DD, Friedman AH, Friedman HS, Vredenburgh JJ (2008) Bevacizumab plus irinotecan in recurrent WHO grade 3 malignant gliomas. Clin Cancer Res 14: 7068–7073
Drevs J, Fakler J, Eisele S, Medinger M, Bing G, Esser N, Marme D, Unger C (2004) Antiangiogenic potency of various chemotherapeutic drugs for metronomic chemotherapy. Anticancer Res 24: 1759–1763
Friedman HS, Prados MD, Wen PY, Mikkelsen T, Schiff D, Abrey LE, Yung WK, Paleologos N, Nicholas MK, Jensen R, Vredenburgh J, Huang J, Zheng M, Cloughesy T (2009) Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol 27(28): 4733–4740
Fulton D, Urtasun R, Forsyth P (1996) Phase II study of prolonged oral therapy with etoposide (VP16) for patients with recurrent malignant glioma. J Neurooncol 27: 149–155
Garcia AA, Hirte H, Fleming G, Yang D, Tsao-Wei DD, Roman L, Groshen S, Swenson S, Markland F, Gandara D, Scudder S, Morgan R, Chen H, Lenz HJ, Oza AM (2008) Phase II clinical trial of bevacizumab and low-dose metronomic oral cyclophosphamide in recurrent ovarian cancer: a trial of the California, Chicago, and Princess Margaret Hospital phase II consortia. J Clin Oncol 26: 76–82
Garcia-Saenz JA, Martin M, Calles A, Bueno C, Rodriguez L, Bobokova J, Custodio A, Casado A, Diaz-Rubio E (2008) Bevacizumab in combination with metronomic chemotherapy in patients with anthracycline- and taxane-refractory breast cancer. J Chemother 20: 632–639
Jain RK, di Tomaso E, Duda DG, Loeffler JS, Sorensen AG, Batchelor TT (2007) Angiogenesis in brain tumours. Nat Rev Neurosci 8: 610–622
Jubb AM, Oates AJ, Holden S, Koeppen H (2006) Predicting benefit from anti-angiogenic agents in malignancy. Nat Rev Cancer 6: 626–635
Kang TY, Jin T, Elinzano H, Peereboom D (2008) Irinotecan and bevacizumab in progressive primary brain tumors, an evaluation of efficacy and safety. J Neurooncol 89: 113–118
Kerbel RS, Kamen BA (2004) The anti-angiogenic basis of metronomic chemotherapy. Nat Rev Cancer 4: 423–436
Kesari S, Schiff D, Doherty L, Gigas DC, Batchelor TT, Muzikansky A, O’Neill A, Drappatz J, Chen-Plotkin AS, Ramakrishna N, Weiss SE, Levy B, Bradshaw J, Kracher J, Laforme A, Black PM, Folkman J, Kieran M, Wen PY (2007) Phase II study of metronomic chemotherapy for recurrent malignant gliomas in adults. Neuro Oncol 9: 354–363
Kreisl TN, Kim L, Moore K, Duic P, Royce C, Stroud I, Garren N, Mackey M, Butman JA, Camphausen K, Park J, Albert PS, Fine HA (2009) Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J Clin Oncol 27: 740–745
Lamborn KR, Yung WK, Chang SM, Wen PY, Cloughesy TF, Deangelis LM, Robins HI, Lieberman FS, Fine HA, Fink KL, Junck L, Abrey L, Gilbert MR, Mehta M, Kuhn JG, Aldape KD, Hibberts J, Peterson PM, Prados MD (2008) Progression-free survival: an important end point in evaluating therapy for recurrent high-grade gliomas. Neuro Oncol 10: 162–170
Macdonald DR, Cascino TL, Schold Jr SC, Cairncross JG (1990) Response criteria for phase II studies of supratentorial malignant glioma. J Clin Oncol 8: 1277–1280
Norden AD, Young GS, Setayesh K, Muzikansky A, Klufas R, Ross GL, Ciampa AS, Ebbeling LG, Levy B, Drappatz J, Kesari S, Wen PY (2008) Bevacizumab for recurrent malignant gliomas: efficacy, toxicity, and patterns of recurrence. Neurology 70: 779–787
Rixe O, Billemont B, Izzedine H (2007) Hypertension as a predictive factor of Sunitinib activity. Ann Oncol 18: 1117
Rubenstein JL, Kim J, Ozawa T, Zhang M, Westphal M, Deen DF, Shuman MA (2000) Anti-VEGF antibody treatment of glioblastoma prolongs survival but results in increased vascular cooption. Neoplasia 2: 306–314
Sathornsumetee S, Cao Y, Marcello JE, Herndon 2nd JE, McLendon RE, Desjardins A, Friedman HS, Dewhirst MW, Vredenburgh JJ, Rich JN (2008) Tumor angiogenic and hypoxic profiles predict radiographic response and survival in malignant astrocytoma patients treated with bevacizumab and irinotecan. J Clin Oncol 26: 271–278
Scartozzi M, Galizia E, Chiorrini S, Giampieri R, Berardi R, Pierantoni C, Cascinu S (2009) Arterial hypertension correlates with clinical outcome in colorectal cancer patients treated with first-line bevacizumab. Ann Oncol 20: 227–230
Schneider BP, Wang M, Radovich M, Sledge GW, Badve S, Thor A, Flockhart DA, Hancock B, Davidson N, Gralow J, Dickler M, Perez EA, Cobleigh M, Shenkier T, Edgerton S, Miller KD (2008) Association of vascular endothelial growth factor and vascular endothelial growth factor receptor-2 genetic polymorphisms with outcome in a trial of paclitaxel compared with paclitaxel plus bevacizumab in advanced breast cancer: ECOG 2100. J Clin Oncol 26: 4672–4678
Scotland RS, Madhani M, Chauhan S, Moncada S, Andresen J, Nilsson H, Hobbs AJ, Ahluwalia A (2005) Investigation of vascular responses in endothelial nitric oxide synthase/cyclooxygenase-1 double-knockout mice: key role for endothelium-derived hyperpolarizing factor in the regulation of blood pressure in vivo. Circulation 111: 796–803
See SJ, Gilbert MR (2004) Anaplastic astrocytoma: diagnosis, prognosis, and management. Semin Oncol 31: 618–634
Stark-Vance V (2005) Bevacizumab and CPT-11 in the treatment of relapsed malignant glioma. In World Federation of Neuro-Oncology Second Quadrennial Meeting and the Sixth Meeting of the European Association for Neuro-Oncology Bigner DD (ed) Vol. 7, pp 369 Duke University Press: Edinburgh, Scotland
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352: 987–996
Twardowski PW, Smith-Powell L, Carroll M, VanBalgooy J, Ruel C, Frankel P, Synold TW (2008) Biologic markers of angiogenesis: circulating endothelial cells in patients with advanced malignancies treated on phase I protocol with metronomic chemotherapy and celecoxib. Cancer Invest 26: 53–59
van den Bent MJ, Vogelbaum MA, Wen PY, Macdonald DR, Chang SM (2009) End point assessment in gliomas: novel treatments limit usefulness of classical Macdonald's criteria. J Clin Oncol 27: 2905–2908
van Heeckeren WJ, Ortiz J, Cooney MM, Remick SC (2007) Hypertension, proteinuria, and antagonism of vascular endothelial growth factor signaling: clinical toxicity, therapeutic target, or novel biomarker? J Clin Oncol 25: 2993–2995
Veronese ML, Mosenkis A, Flaherty KT, Gallagher M, Stevenson JP, Townsend RR, O′Dwyer PJ (2006) Mechanisms of hypertension associated with BAY 43–9006. J Clin Oncol 24: 1363–1369
Vredenburgh JJ, Desjardins A, Herndon 2nd JE, Dowell JM, Reardon DA, Quinn JA, Rich JN, Sathornsumetee S, Gururangan S, Wagner M, Bigner DD, Friedman AH, Friedman HS (2007a) Phase II trial of bevacizumab and irinotecan in recurrent malignant glioma. Clin Cancer Res 13: 1253–1259
Vredenburgh JJ, Desjardins A, Herndon 2nd JE, Marcello J, Reardon DA, Quinn JA, Rich JN, Sathornsumetee S, Gururangan S, Sampson J, Wagner M, Bailey L, Bigner DD, Friedman AH, Friedman HS (2007b) Bevacizumab plus irinotecan in recurrent glioblastoma multiforme. J Clin Oncol 25: 4722–4729
Wen PY, Kesari S (2008) Malignant gliomas in adults. N Engl J Med 359: 492–507
Wen PY, Schiff D, Kesari S, Drappatz J, Gigas DC, Doherty L (2006) Medical management of patients with brain tumors. J Neurooncol 80: 313–332
Wong ET, Hess KR, Gleason MJ, Jaeckle KA, Kyritsis AP, Prados MD, Levin VA, Yung WK (1999) Outcomes and prognostic factors in recurrent glioma patients enrolled onto phase II clinical trials. J Clin Oncol 17: 2572–2578
Yang R, Ogasawara AK, Zioncheck TF, Ren Z, He GW, DeGuzman GG, Pelletier N, Shen BQ, Bunting S, Jin H (2002) Exaggerated hypotensive effect of vascular endothelial growth factor in spontaneously hypertensive rats. Hypertension 39: 815–820
Yung WK, Albright RE, Olson J, Fredericks R, Fink K, Prados MD, Brada M, Spence A, Hohl RJ, Shapiro W, Glantz M, Greenberg H, Selker RG, Vick NA, Rampling R, Friedman H, Phillips P, Bruner J, Yue N, Osoba D, Zaknoen S, Levin VA (2000) A phase II study of temozolomide vs procarbazine in patients with glioblastoma multiforme at first relapse. Br J Cancer 83: 588–593
Zuniga RM, Torcuator R, Jain R, Anderson J, Doyle T, Ellika S, Schultz L, Mikkelsen T (2009) Efficacy, safety and patterns of response and recurrence in patients with recurrent high-grade gliomas treated with bevacizumab plus irinotecan. J Neurooncol 91: 329–336
Acknowledgements
This work was supported by the NIH Grants 5P50-NS-20023 and 5 R37 CA11898; the NIH Grant MO1 RR 30, GCRC Program, NCRR; and the NCI SPORE 1 P20 CA096890; and a grant from Genentech Pharmaceuticals.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on British Journal of Cancer website (http://www.nature.com/bjc)
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Reardon, D., Desjardins, A., Vredenburgh, J. et al. Metronomic chemotherapy with daily, oral etoposide plus bevacizumab for recurrent malignant glioma: a phase II study. Br J Cancer 101, 1986–1994 (2009). https://doi.org/10.1038/sj.bjc.6605412
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6605412
Keywords
This article is cited by
-
Clinical activity and safety of sintilimab, bevacizumab, and TMZ in patients with recurrent glioblastoma
BMC Cancer (2024)
-
Glioma targeted therapy: insight into future of molecular approaches
Molecular Cancer (2022)
-
Ribosomal protein S11 influences glioma response to TOP2 poisons
Oncogene (2020)
-
Correlation of radiological and immunochemical parameters with clinical outcome in patients with recurrent glioblastoma treated with Bevacizumab
Clinical and Translational Oncology (2019)
-
Phase I/II trial of vorinostat, bevacizumab, and daily temozolomide for recurrent malignant gliomas
Journal of Neuro-Oncology (2018)

{kind=link}