
Abstract
Background:
Resistance to trastuzumab is often observed in women with human epidermal growth factor receptor 2 (HER2)-positive breast cancer and has been shown to involve multiple potential mechanisms. We examined the ability of microarray analyses to determine the potential markers of pathological complete response (pCR).
Methods:
We conducted an analysis of tumours from 38 patients with locally advanced HER2-positive breast cancer who had received trastuzumab combined with docetaxel. Quantitative reverse transcriptase (RT)–PCR was used to assess the expression of 30 key genes; microarray analyses were carried out on 25 tumours to identify a prognostic gene expression profile, with 13 blinded samples used to validate the identified profile.
Results:
No gene was found to correlate with response by RT–PCR. The microarray analysis identified a gene expression profile of 28 genes, with 12 upregulated in the pCR group and 16 upregulated in non-pCR. The leave-one-out cross-validation test exhibited 72% accuracy, 86% specificity, and 55% sensitivity. The 28-gene expression profile classified the 13 validation samples with 92% accuracy, 89% specificity, and 100% sensitivity.
Conclusion:
Our results suggest that genes not involved in classical cancer pathways such as apoptosis or DNA repair could be involved in responses to a trastuzumab–docetaxel-based regimen. They also describe for the first time a gene expression signature that predicts trastuzumab response.
Similar content being viewed by others

Main
Amplification and overexpression of human epidermal growth factor receptor 2 (HER2) is observed in 20–30% of invasive breast cancer (Slamon et al, 1987) and correlates with tumour progression and poor prognosis. Although the EGFR family stimulates mitogenesis through ligand-induced pathways, there is no known ligand for HER2. Increased HER2 expression induces a signalling pathway that involves Ras and Src, as well as PI3K/Akt, and is associated with tumour formation (Siegel et al, 1994).
Trastuzumab (Herceptin; F Hoffmann-La Roche, Basel, Switzerland) is a humanised monoclonal antibody directed against the HER2 protein. It produces significant (>50%) tumour regression in ∼15% of patients with HER2-positive metastatic breast cancer that is refractory to conventional therapy, and in ∼23% of patients when used as first-line therapy (Cobleigh et al, 1999). The addition of trastuzumab to standard chemotherapy significantly improves the response rate, response duration, and survival. The clinical benefits of trastuzumab-based therapies have been well documented in both adjuvant (Joensuu et al, 2006) and metastatic settings (Marty et al, 2005). However, the precise molecular pathways through which trastuzumab exerts its anti-tumour effects in breast cancer cells are not yet fully understood. Trastuzumab action involves multiple mechanisms, including the induction of apoptotic signalling pathways, cell cycle perturbation, and cellular cytotoxicity (Sliwkowski et al, 1999). Treatment with trastuzumab dephosphorylates and downregulates HER2, leading to significant clinical efficacy against HER2-positive breast cancer. It also sensitises breast cancer cells to chemotherapeutic agents, especially to tubulin-polymerising agents and radiation therapy (Baselga et al, 1998; Liang et al, 2003). It was shown that anti-HER2 monoclonal antibodies inhibit HER2-overexpressing breast cancer cells through G1 cell cycle arrest, which was associated with the induction of the cyclin-dependent kinase (CDK) inhibitor p27kip1 and reduction of CDK2 (Le et al, 2003). Trastuzumab may also inhibit the PI3K/Akt pathway by promoting PTEN activation (Nagata et al, 2004). Trastuzumab has been shown to reduce tumour volume and microvessel density in HER2-positive breast cancer models in vivo (Laughner et al, 2001; Izumi et al, 2002). Synergy with DNA-damaging drugs is thought to be due to trastuzumab-mediated inhibition of DNA repair. Trastuzumab partially inhibits the repair of DNA adducts in vitro after treatment with cisplatin and blocks unscheduled DNA synthesis after radiation (Pietras et al, 1994, 1999). Finally, trastuzumab has also been shown to be associated with immunoreactive actions through antibody-directed cellular cytotoxicity (ADCC) (Arnould et al, 2006).
Recently, trastuzumab-based neoadjuvant chemotherapy has been shown to achieve promising efficacy, with a good pathological complete response (pCR) rate, while being well tolerated in women with stage II or III HER2-positive breast cancer (Buzdar et al, 2005; Coudert et al, 2006, 2007). Among taxanes, docetaxel associated with trastuzumab shows evidence of improved efficacy in obtaining pCR rates.
In this study, we examined the expression of a panel of 30 genes involved in cell cycle progression, DNA repair, and apoptosis, which may have a putative role in trastuzumab resistance, in a series of breast carcinomas that had been treated with trastuzumab-based neoadjuvant chemotherapy. In parallel, we used microarray analysis on the same tumour samples to identify a potential marker of pCR that may have a prognostic value in identifying patients who are more likely to respond to trastuzumab therapy.
Materials and methods
Patients and samples
We retrospectively studied a population of 38 patients who had received trastuzumab in combination with chemotherapy as primary systemic therapy for their operable, HER2-positive, stage II/III breast cancer (Table 1). All patients provided written, informed consent for their tissue material and clinical data to be used for research purposes. Patients were treated in two open-label phase II clinical trials: TAXHER01 (n=29) and GETNA01 (n=9) (Coudert et al, 2006, 2007).
All patients received weekly neoadjuvant trastuzumab (4 mg kg−1 loading dose, followed by 2 mg kg−1 once weekly) in combination with either docetaxel alone (100 mg m−2 every 3 weeks for six cycles) or docetaxel (75 mg m−2 every 3 weeks for six cycles) combined with carboplatin (AUC 6) every 3 weeks for six cycles. The pCR rates were assessed using Chevallier's classification (Chevallier et al, 1993) 3 weeks after the last course of trastuzumab-containing neoadjuvant treatment. An absence of disease in the breast or in the lymph nodes, with or without in situ carcinoma, was considered to be a pCR. The HER2 status was determined using both immunohistochemistry and fluorescence in situ hybridisation (Coudert et al, 2006, 2007).
HER-2 testing
The HER-2 status was analysed before treatment in each tumour according to ASCO guidelines for immunohistochemistry (IHC) or fluorescent in situ hybridisation (FISH) (Wolff et al, 2007) IHC was carried out with an anti-HER2 antibody (clone 4B5) on a Ventana Benchmark XT automate (Ventana Medical Systems, Tucson, AZ, USA). All tumours were considered as positive if >30% of tumour cells display a complete and strongly positive membrane staining. As described in a previous report (Coudert et al, 2006, 2007; Arnould et al, 2006), all biopsies were also retrospectively analysed with FISH procedures that confirm that all tumours included in this study displayed an HER2 gene amplification with a mean of more than six copies of the HER2 gene.
RNA extraction
Needle core biopsy samples were taken at baseline, with one used for the initial diagnosis and two used for RNA extraction. All tissue samples were snap frozen and stored in liquid nitrogen, and only samples containing ⩾30% tumour cells were analysed further. Total RNA was extracted from tissue samples by using the TRIzol method as recommended by the manufacturer (Invitrogen Corporation, Carlsbad, CA, USA). The quantity, quality, and purity of extracted RNA were assessed using a NanoDrop 1000 spectrophotometer (NanoDrop, Wilmington, DE, USA) at 260 and 280 nm (the A260/280 ratio of pure RNA is higher than 1.8) and an Agilent 2100 bioanalyser (Agilent, Santa Clara, CA, USA). Total RNA from a pool of four normal mammary tissues was used as normal sample, and RNA extracted from the MCF-7 human breast cancer cell line was used to calibrate real-time quantitative and reverse transcriptase (RT)–PCR.
RT–PCR and real-time quantitative PCR
One microgram of total RNA was reverse transcribed in 20 μl of RT–PCR. Real-time quantitative PCR was carried out on an ABI PRISM 7300 (Applied Biosystems, Foster City, CA, USA) using the TaqMan method. Analysis of 18S ribosomal RNA was used to assess complementary DNA (cDNA) quality and as a reference control. Results were analysed at the Ct level and references for the genes analysed are summarised in Table 2. Survivin, caspase-3, and their splice variant expressions were determined by design primers and probes labelled at the 5′ end with FAM and at the 3′ end with TAMRA. Assays on Demand (Applied Biosystems) were used for the other studied genes. The results were analysed using either the 2−ΔCt method for expression comparison or the 2−ΔΔCt method (Vegran et al, 2007) for statistical analyses.
Statistical analyses were carried out with Statview 5.0 software (SAS Institute, Inc., Cary, NC, USA). The non-parametric Mann–Whitney U-test was used to compare gene expression with pathological response. Statistical significance was considered when P-value was <0.05.
Microarray experiment
Microarray analyses were carried out using the Affymetrix-Microarray Platform of the Institute of Genetics and Molecular and Cellular Biology (IGBMC) and Génopole Alsace-Lorraine (Dr Philippe Kastner). The analysis used samples from 25 patients (11 with pCR and 14 with non-pCR) who constituted the training set. It was randomly constituted with the 25 first patients enrolled in the study. The resulting profile was validated using an independent and blinded group of 13 patients (four with pCR and nine with non-pCR) belonging to the test set. It was also randomly constituted with the 13 patients who joined the protocol after the beginning of training set microarray analysis.
The fluorescent nucleic acids hybridised onto the microarrays were prepared from total RNA. One microgram of total RNA was reverse transcribed into cDNA using a poly-dT with an extended region as a 3′ end primer. After second-strand synthesis, all the different double-strand cDNAs had a common 3′ end extension, which was used as a specific annealing site during PCR amplification. This unidirectional PCR amplification produced single-strand linear PCR products, which were labelled by random priming with dUTP-Cy5 (red) for the test samples or with dUTP-Cy3 (green) for the reference samples. Test and reference samples were co-hybridised onto microarrays. Human microarrays from the Affymetrix-Microarray Platform of the IGBMC and Génopole Alsace-Lorraine were used, onto which 25 000 genes were spotted. Reference genes were eliminated. Hybridised slides were scanned to detect fluorescence signals at high resolution. Fluorescent intensities were normalised and standardised using IGBMC in-house ‘Elea’ software, followed by a LOcal Weighted Estimates of Smooth Scatterplots (LOWESS) fitting-based method. Briefly, genes were selected as invariants from ranks of values in the Cy3 and Cy5 channels, and were then used in the LOWESS algorithm to compute the normalisation factor between the two channel values. This generated two values: the signal value A=Log2 (test value * reference value)/2 and the log ratio M=Log2 (test value/reference value).
Microarray data analysis
Using A values, we determined the lowest median expression level of the population and excluded every gene with an A value lower than this. Using this heuristic filtering, we identified 14 829 genes for further analysis. From this subset of genes, statistical filtering was performed on M values using IGBMC in-house statistical ‘Zoe’ software. The Mann–Whitney U-test was then used with 1000 permutations to compare pCR and non-pCR rates. Only genes with P<0.002 were kept in the signature to discriminate pCR and non-pCR groups. P-values and q-values (for false discovery rate) were presented in Table 3.
The classification of patients constituting the test set was performed with the calculation of the correlation coefficient between the microarray values of each test patient and the mean of the 28-gene microarray expression value of pCR and non-pCR determined with the training set. A patient was classified as a pCR when the correlation coefficient obtained with the mean training pCR values was superior to the one obtained with the mean training non-pCR values, and inversely. As mentioned above, the classification of patients belonging to the test set was blindly performed. The comparison with real patient response was carried out later.
Leave-one-out cross-validation test
The leave-one-out cross-validation test was performed on the training set patients. One patient was randomly suppressed. On the new 24-patient training set, a Mann–Whitney U-test was carried out with 1000 permutations to compare pCR and non-pCR rates. Only genes with P<0.002 were kept to generate a signature discriminating pCR and non-pCR groups. Thereafter, the excluded patient was classified with the calculation of the correlation coefficient between the microarray values of the patient and the mean of the gene microarray expression value of pCR and non-pCR determined with the 24-patient training set. One patient was excluded each time, generating 25 different tests.
Results
Analysis of selected gene expression by quantitative RT–PCR
When the relative expression of genes associated with cell cycle progression was compared with pathological response, it was found that the expression of these genes did not correlate with the observed pathological response. We next compared the relative expression of DNA repair genes with pathological response, and the results similarly showed that the expression of these genes did not correlate with pathological response. No relationship was found with the relative expression of apoptotic genes either.
Microarray data analysis
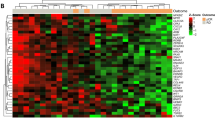
Of the 25 patients in the training set, 11 (44%) showed pCR and 14 (56%) had non-pCR. Microarray analysis of tumour samples from these patients indicated that expression significantly differed between pCR and non-pCR tumour samples for 28 genes (Figure 1A). Among these 28 genes, 12 were more highly expressed in pCR tumour samples (WEE1, ZNF146, SENP7, GPR22, KIAA1549, SYNCRIP, SLC30A6, GRHL2, CCDC123, LOC340171, STX1A, cDNA FLJ11973 fis, and clone HEMBB1001221), and 16 genes were highly expressed in non-pCR samples (LOC158402, PITPNA, PPP2CA, SLC35A4, NFE2L1, C5orf3, PEX19, P2RX1, CDC14A, SENP8, PSMD11, CTNS, DER1, PRKACA, LAMA3, and FLJ20160) (Table 4). In addition, there was no difference observed for treatment effect (TAXHER01 or GETNA01) on this 28-gene expression profile.

Hierarchical clustering of the 28 genes discriminating both pathological complete response (pCR) and non-pathological complete response (non-pCR) for the 25 patient training (A) and 13 patient test (B) sets. Green and red colours represent underexpression or overexpression centred to median array values, respectively.
The discriminatory 28-gene profile was validated using a leave-one-out cross-validation test. The analysis of the profile was carried out without previous knowledge of the patients' pathological response. One patient was excluded each time and was classified using a correlation coefficient based on the mean expression value of each selected gene for the pCR and non-pCR subsets. A patient was classified as being in the pCR group when their correlation coefficient was higher, with mean values above the non-pCR values, and vice versa. Using this approach, the leave-one-out cross-validation test classified 6 out of 11 pCR patients as having the pCR expression profile, and 12 out of 14 non-pCR patients into the non-pCR profile. Thus, the gene expression profile exhibited 55% sensitivity, 86% specificity, and 72% accuracy (Table 5).
To proceed further, the discriminatory 28-gene profile was then validated using the independent cohort of 13 patients. Analysis of the profile was carried out without earlier knowledge of the patients' pathological response. Each patient's tumour sample was classified using a correlation coefficient based on the mean expression value of each selected gene for the pCR and non-pCR subsets. A patient was classified as being in the pCR group when their correlation coefficient was higher, with mean values above the non-pCR values, and vice versa. Using this approach, our 28-gene profile correctly classified the four pCR patients as having the pCR expression profile, and 8 out of 9 non-pCR patients into the non-pCR profile (Figure 1B). Thus, our 28-gene profile for a trastuzumab–docetaxel-based regimen exhibited 100% sensibility, 89% specificity, and 92% accuracy (Table 6).
Discussion
The aim of oncology is to provide the most appropriate cancer treatment to ensure the best patient response. However, it is very difficult to choose the best combination of chemotherapy agents, and it is necessary to develop new tools that will aid in making the best treatment choice. In this study, we explored gene expression profiles to predict response to trastuzumab–docetaxel-based chemotherapy in women with locally advanced HER2-positive breast cancer.
The real-time quantitative PCR study on 30 genes involved in cell cycle progression, DNA repair, or apoptosis revealed that these genes did not seem to be predictive for pathological response. Trastuzumab-induced apoptosis has been demonstrated in both breast tumour cell lines and breast carcinomas (Brodowicz et al, 2001; Milella et al, 2004; Emi et al, 2005; Henson et al, 2006). However, in our study, we failed to highlight a role for apoptosis-related genes in our response discriminating profile. This could be explained by immunoreactive actions through ADCC (Arnould et al, 2006).
Using microarray analysis, we generated a 28-gene profile that could discriminate between tumour samples that would attain a pCR and those that would not in response to treatment with a trastuzumab–docetaxel-based regimen. This profile was not affected by treatment effect (TAXHER01 or GETNA01), and the results confirm previous analyses that have commented on the association between pCR and HER2 amplification (Arnould et al, 2006; Coudert et al, 2006, 2007). In addition, the expression values of the 30 selected genes analysed with real-time quantitative PCR were concordant with those that overlapped with high-throughput microarray, confirming the absence of involvement of these genes.
In the leave-one-out cross-validation test, the classifier shows 72% accuracy, 86% specificity, and 55% sensitivity. The 28-gene expression profile classified the 13 test samples with 92% accuracy, 89% specificity, and 100% sensitivity. The performance with the test set is better than that with the training set, conforming previous observations showing that independent validation is the gold standard to evaluate the performance of the prediction rule (Michiels et al, 2007). However, the main characteristic of this classifier is high specificity, both with training and test sets, allowing the identification of patients resistant to trastuzumab–docetaxel-based treatment.
Among the genes identified in the profile, NFE2L1 was upregulated in the non-pCR group. NFE2L1 has been described to be a regulator of detoxifying enzyme expression, and, in association with Jun, is able to induce the expression of genes encoding detoxifying enzymes. As a result, overexpression of NFE2L1 could protect tumour cells by decreasing the toxicity of treatment. Moreover, overexpression of NFE2L1 was described as having the same impact as c-Myc overexpression (Morrish et al, 2003), suggesting that this gene could be implicated in resistance to chemotherapy.
Two small ubiquitin-like (SUMO)/sentrin-specific protease (SENP) family members were differentially expressed in both pCR and non-pCR groups. Thus, SENP7 was upregulated in the pCR group, whereas SENP8 was strongly expressed in the non-pCR group. Although the properties and targets of SENP7 have not yet been determined, it has been established that SENP8 is an NEDD8-, rather than SUMO-, specific protease (Hay, 2007).
The WEE1 gene harbours a large expression level in the pCR group. This gene suppresses the activity of the Cyclin B1–Cdc2 complex, suggesting its implication in the response process to trastuzumab–docetaxel-based treatments (Yoshida et al, 2004). The GRHL2 gene, which is overexpressed in the pCR group as well, is involved in the regulation of hTERT. The GRHL2 downregulation by siRNA induced a decrease in hTERT activity and increased the immortalisation process (Kang et al, 2009).
PPP2CA is described as an anti-apoptotic gene (Hu et al, 2009), which could explicate why it is overexpressed in the non-pCR group. Other genes are overexpressed in the non-pCR set. For example, CDC14A is able to interact with and inhibit p53 and the Cyclin B–Cdk1 complex (Paulsen et al, 2006), and PSMD11 has been found to be overexpressed in breast carcinomas (Deng et al, 2007).
Surprisingly, four genes that discriminated between responses to treatment have been described as being involved in either synaptic transmission (SYNCRIP, P2RX1, and STX1A) or brain development (KLHL2). These results could highlight a new role of these genes in breast cancer.
Several studies have analysed gene expression profiles of breast carcinomas treated with docetaxel-based regimens. A 92-gene profile was identified that discriminated between docetaxel-resistant and -sensitive breast carcinomas (Chang et al, 2003). The functional classes of these differentially expressed genes were apoptosis, cell adhesion or cytoskeleton, protein transport, signal transduction, RNA transcription, RNA splicing or transport, cell cycle, and protein translation. Further, a 512-gene signature was described as predicting pCR to primary systemic therapy with gemcitabine, epirubicin, and docetaxel (Thuerigen et al, 2006). This signature contained a predominance of genes encoding enzymes and proteins binding to nucleic acids, many of which were transcriptional regulators. Another study on 44 breast tumour tissues identified 85 genes that predicted a clinical response to docetaxel with 80% accuracy (Iwao-Koizumi et al, 2005). The most prominent characteristic in non-responders was the elevated expression of genes controlling the cellular redox environment (glutathione and thioredoxin systems). Lastly, an in vitro study recently identified 50 genes involved in docetaxel sensitivity that were able to predict the response in 22 out of 24 clinical samples that were used in Chang's study (Potti et al, 2006).
To date, only one study has used RNA profiling to predict responses to trastuzumab–vinorelbine-based treatments in patients with early HER2-positive breast cancer (Harris et al, 2007). In this study, resistant tumours exhibited a higher expression of several growth factors, growth factor receptors, the PI3K regulatory subunit p85, microtubule-associated protein 2, and some basal genes. Although the chemotherapeutic agent used with trastuzumab is different, this signature was not confirmed in an independent set of patients to validate the identified profile. In addition, no predictive genes were identified in pCR tumours.
In conclusion, our results suggest that genes not involved in classical cancer pathways, such as apoptosis, cell cycle progression, or DNA repair, could be involved in determining responses to a trastuzumab–docetaxel-based regimen. Importantly, our results identify for the first time a gene expression signature that predicts trastuzumab response in breast carcinoma. A consequence of individualised treatment is that it can be difficult to identify appropriate numbers of patients with similar characteristics who have been exposed to the same treatment regimen to adequately statistically power the study. Thus, the prognostic accuracy of the 28-gene profile that we identified will be confirmed in a new multi-centre cohort of patients using a multivariate analysis in a larger number of cases.
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Arnould L, Gelly M, Penault-Llorca F, benoit L, Bonnetain F, Migoen C, Cabaret V, fermeaux V, Bertheau P, Garnier J, Jeanin JF, Coudert B (2006) Trastuzumab-based treatment of HER2-positive breast cancer: an antibody-dependent cellular cytotoxicity mechanism? Br J Cancer 94: 259–267
Baselga J, Norton L, Albanell J, Kim YM, Mendelsohn J (1998) Recombinant humanized anti-HER2 antibody (Herceptin) enhances the antitumor activity of paclitaxel and doxorubicin against HER2/neu overexpressing human breast cancer xenografts. Cancer Res 58: 2825–2831
Brodowicz T, Kandioler D, Tomek S, Ludwig C, Rudas M, Kunstfeld R, Koestler W, Hejna M, Budinsky A, Wiltschke C, Zielinski CC (2001) Anti-Her-2/neu antibody induces apoptosis in Her-2/neu overexpressing breast cancer cells independently from p53 status. Br J Cancer 85: 1764–1770
Buzdar AU, Ibrahim NK, Francis D, Booser DJ, Thomas ES, Theriault RL, Pusztai L, Green MC, Arun BK, Giordano SH, Cristofanilli M, Frye DK, Smith TL, Hunt KK, Singletary SE, Sahin AA, Ewer MS, Buchholz TA, Berry D, Hortobagyi GN (2005) Significantly higher pathologic complete remission rate after neoadjuvant therapy with trastuzumab, paclitaxel, and epirubicin chemotherapy: results of a randomized trial in human epidermal growth factor receptor 2-positive operable breast cancer. J Clin Oncol 23: 3676–3685
Chang JC, Wooten EC, Tsimelzon A, Hilsenbeck SG, Gutierrez MC, Elledge R, Mohsin S, Osborne CK, Chamness GC, Allred DC, O'Connell P (2003) Gene expression profiling for the prediction of therapeutic response to docetaxel in patients with breast cancer. Lancet 362: 362–369
Chevallier B, Roche H, Olivier JP, Chollet P, Hurteloup P (1993) Inflammatory breast cancer. Pilot study of intensive induction chemotherapy (FEC-HD) results in a high histologic response rate. Am J Clin Oncol 16: 223–228
Cobleigh MA, Vogel CL, Tripathy D, Robert NJ, Scholl S, Fehrenbacher L, Wolter JM, Paton V, Shak S, Lieberman G, Slamon DJ (1999) Multinational study of the efficacy and safety of humanized anti-HER2 monoclonal antibody in women who have HER2-overexpressing metastatic breast cancer that has progressed after chemotherapy for metastatic disease. J Clin Oncol 17: 2639–2648
Coudert BP, Arnould L, Moreau L, Chollet P, Weber B, Vanlemmens L, Molucon C, Tubiana N, Causeret S, Misset JL, Feutray S, Mery-Mignard D, Garnier J, Fumoleau P (2006) Pre-operative systemic (neo-adjuvant) therapy with trastuzumab and docetaxel for HER2-overexpressing stage II or III breast cancer: results of a multicenter phase II trial. Ann Oncol 17: 409–414
Coudert BP, Largillier R, Arnould L, Chollet P, Campone M, Coeffic D, Priou F, Gligorov J, Martin X, Trillet-Lenoir V, Weber B, Bleuse JP, Vasseur B, Serin D, Namer MJ (2007) Multicenter phase II trial of neoadjuvant therapy with trastuzumab, docetaxel, and carboplatin for human epidermal growth factor receptor-2-overexpressing stage II or III breast cancer: results of the GETN(A)-1 trial. J Clin Oncol 25: 2678–2684
Deng S, Zhou H, Xiong R, Lu Y, Yan D, Xing T, Dong L, Tang E, Yang H (2007) Over-expression of genes and proteins of ubiquitin specific peptidases (USPs) and proteasome subunits (PSs) in breast cancer tissue observed by the methods of RFDD-PCR and proteomics. Breast Cancer Res Treat 104: 21–30
Emi M, Kim R, Tanabe K, Uchida Y, Toge T (2005) Targeted therapy against Bcl-2-related proteins in breast cancer cells. Breast Cancer Res 7: R940–R952
Harris LN, You F, Schnitt SJ, Witkiewicz A, Lu X, Sgroi D, Ryan PD, Come SE, Burstein HJ, Lesnikoski BA, Kamma M, Friedman PN, Gelman R, Iglehart JD, Winer EP (2007) Predictors of resistance to preoperative trastuzumab and vinorelbine for HER2-positive early breast cancer. Clin Cancer Res 13: 1198–1207
Hay RT (2007) SUMO-specific proteases: a twist in the tail. Trends Cell Biol 17: 370–376
Henson ES, Hu X, Gibson SB (2006) Herceptin sensitizes ErbB2-overexpressing cells to apoptosis by reducing antiapoptotic Mcl-1 expression. Clin Cancer Res 12: 845–853
Hu X, Wu X, Xu J, Zhou J, Han X, Guo J (2009) Src Kinase up-regulates the ERK cascade through inactivation of protein phosphatase 2A following cerebral ischemia. BMC Neurosci 14: 10–74
Iwao-Koizumi K, Matoba R, Ueno N, Kim SJ, Ando A, Miyoshi Y, Maeda E, Noguchi S, Kato K (2005) Prediction of docetaxel response in human breast cancer by gene expression profiling. J Clin Oncol 23: 422–431
Izumi Y, Xu L, di Tomaso E, Fukumura D, Jain RK (2002) Tumour biology: Herceptin acts as an anti-angiogenic cocktail. Nature 416: 279–280
Joensuu H, Kellokumpu-Lehtinen PL, Bono P, Alanko T, Kataja V, Asola R, Utriainen T, Kokko R, Hemminki A, Tarkkanen M, Turpeenniemi-Hujanen T, Jyrkkiö S, Flander M, Helle L, Ingalsuo S, Johansson K, Jääskeläinen AS, Pajunen M, Rauhala M, Kaleva-Kerola J, Salminen T, Leinonen M, Elomaa I, Isola J (2006) Adjuvant docetaxel or vinorelbine with or without trastuzumab for breast cancer. N Engl J Med 354: 809–820
Kang X, Chen W, Kim RH, Kang MK, Park NH (2009) Regulation of the hTERT promoter activity by MSH2, the hnRNPs K and D, and GRHL2 in human oral squamous cell carcinoma cells. Oncogene 28: 565–574
Laughner E, Taghavi P, Chiles K, Mahon PC, Semenza GL (2001) HER2 (neu) signalling increases the rate of hypoxia-inducible factor 1alpha (HIF-1alpha) synthesis: novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol Cell Biol 21: 3995–4004
Le XF, Claret FX, Lammayot A, Tian L, Deshpande D, LaPushin R, Tari AM, Bast Jr RC (2003) The role of cyclin-dependent kinase inhibitor p27Kip1 in anti-HER2 antibody-induced G1 cell cycle arrest and tumor growth inhibition. J Biol Chem 278: 23441–23450
Liang K, Lu Y, Jin W, Ang KK, Milas L, Fan Z (2003) Sensitization of breast cancer cells to radiation by trastuzumab. Mol Cancer Ther 2: 1113–1120
Marty M, Cognetti F, Maraninchi D, Snyder R, Mauriac L, Tubiana-Hulin M, Chan S, Grimes D, Anton A, Lluch A, Kennedy J, O'Byrne K, Cont P, Green M, Ward C, Mayne K, Extra JM (2005) Randomized phase II trial of the efficacy and safety of trastuzumab combined with docetaxel in patients with human epidermal growth factor receptor 2–positive metastatic breast cancer administered as first-line treatment: the M77001 study group. J Clin Oncol 23: 4265–4274
Michiels S, Koscielny S, Hill C (2007) Interpretation of microarray data in cancer. Br J Cancer 96: 1155–1158
Milella M, Trisciuoglio D, Bruno T, Ciuffreda L, Mottolese M, Cianciulli A, Cognetti F, Zangemeister-Wittke U, Del Bufalo D, Zupi G (2004) Trastuzumab down-regulates Bcl-2 expression and potentiates apoptosis induction by Bcl-2/Bcl-XL bispecific antisense oligonucleotides in HER-2 gene – amplified breast cancer cells. Clin Cancer Res 10: 7747–7756
Morrish F, Giedt C, Hockenbery D (2003) c-MYC apoptotic function is mediated by NRF-1 target genes. Genes Dev 17: 240–255
Nagata Y, Lan K-H, Zhou X, Tan M, Esteva FJ, Sahin AA, Klos KS, Li P, Monia BP, Nguyen NT, Hortobagyi GN, Hung MC, Yu D (2004) PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell 6: 117–127
Paulsen MT, Starks AM, Derheimer FA, Hanasoge S, Li L, Dixon JE, Ljungman M (2006) The p53-targeting human phosphatase hCdc14A interacts with the Cdk1/cyclin B complex and is differentially expressed in human cancers. Mol Cancer 5: 25
Pietras RJ, Fendly BM, Chazin VR, Pegram MD, Howell SB, Slamon DJ (1994) Antibody to HER-2/neu receptor blocks DNA repair after cisplatin in human breast and ovarian cancer cells. Oncogene 9: 1829–1838
Pietras RJ, Poen JC, Gallardo D, Wongvipat PN, Lee HJ, Slamon DJ (1999) Monoclonal antibody to HER-2/neu receptor modulates repair of radiation-induced DNA damage and enhances radiosensitivity of human breast cancer cells overexpressing this oncogene. Cancer Res 59: 1347–1355
Potti A, Dressman HK, Bild A, Riedel RF, Chan G, Sayer R, Cragun J, Cottrill H, Kelley MJ, Petersen R, Harpole D, Marks J, Berchuck A, Ginsburg GS, Febbo P, Lancaster J, Nevins JR (2006) Genomic signatures to guide the use of chemotherapeutics. Nat Med 12: 1294–1300
Siegel PM, Dankort DL, Hardy WR, Muller WJ (1994) Novel activating mutations in the neu proto-oncogene involved in induction of mammary tumors. Mol Cell Biol 14: 7068–7077
Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL (1987) Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 235: 177–182
Sliwkowski MX, Lofgren JA, Lewis GD, Hotaling TE, Fendly BM, Fox JA (1999) Nonclinical studies addressing the mechanism of action of trastuzumab (Herceptin). Semin Oncol 26 (Suppl 12): 60–70
Thuerigen O, Schneeweiss A, Toedt G, Warnat P, Hahn M, Kramer H, Brors B, Rudlowski C, Benner A, Schuetz F, Tews B, Eils R, Sinn HP, Sohn C, Lichter P (2006) Gene expression signature predicting pathologic complete response with gemcitabine, epirubicin, and docetaxel in primary breast cancer. J Clin Oncol 24: 1839–1845
Vegran F, Boidot R, Oudin C, Defrain C, Rebucci M, Lizard-Nacol S (2007) Association of p53 gene alterations with the expression of antiapoptotic survivin splice variants in breast cancer. Oncogene 26: 290–297
Wolff AC, Hammond ME, Schwartz JN, Hagerty KL, Allred DC, Cote RJ, Dowsett M, Fitzgibbons PL, Hanna WM, Langer A, McShane LM, Paik S, Pegram MD, Perez EA, Press MF, Rhodes A, Sturgeon C, Taube SE, Tubbs R, Vance GH, van de Vijver M, Wheeler TM, Hayes DF (2007) American Society of Clinical Oncology; College of American Pathologists. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. J Clin Oncol 25: 118–145
Yoshida T, Tanaka S, Mogi A, Shitara Y, Kuwano H (2004) The clinical significance of Cyclin B1 and Wee1 expression in non-small-cell lung cancer. Ann Oncol 15: 252–256
Acknowledgements
This work was supported by the Conseil Régional de Bourgogne and by the Association pour la Recherche sur le Cancer. We thank Bernard Jost and Christine Bole-Feysot from Plateforme Biopuces de Strasbourg for microarray experiment implementation, and Berangère Vasseur from Roche Laboratory, France.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Végran, F., Boidot, R., Coudert, B. et al. Gene expression profile and response to trastuzumab–docetaxel-based treatment in breast carcinoma. Br J Cancer 101, 1357–1364 (2009). https://doi.org/10.1038/sj.bjc.6605310
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6605310
Keywords
This article is cited by
-
Accurate prediction of breast cancer survival through coherent voting networks with gene expression profiling
Scientific Reports (2021)
-
Genes and functions from breast cancer signatures
BMC Cancer (2018)
-
Transcriptional expression of 8 genes predicts pathological response to first-line docetaxel + trastuzumab-based neoadjuvant chemotherapy
BMC Cancer (2015)
-
PRKACA mediates resistance to HER2-targeted therapy in breast cancer cells and restores anti-apoptotic signaling
Oncogene (2015)
-
Defining the molecular response to trastuzumab, pertuzumab and combination therapy in ovarian cancer
British Journal of Cancer (2012)
