
Abstract
The HOX genes are a family of homeodomain-containing transcription factors that determine the identity of cells and tissues during embryonic development. They are also known to behave as oncogenes in some haematological malignancies. In this study, we show that the expression of many of the HOX genes is highly elevated in primary non-small-cell lung cancers (NSCLCs) and in the derived cell lines A549 and H23. Furthermore, blocking the activity of HOX proteins by interfering with their binding to the PBX co-factor causes these cells to undergo apoptosis in vitro and reduces the growth of A549 tumours in vivo. These findings suggest that the interaction between HOX and PBX proteins is a potential therapeutic target in NSCLC.
Similar content being viewed by others


Main
The HOX genes are a family of homeodomain-containing transcription factors that determine the identity of cells and tissues in early development (Iimura and Pourquié, 2007), and also have key regulatory roles in adult haematopoietic stem cells and their descendants (Abramovich and Humphries, 2005). In addition, HOX genes are often overexpressed in malignant cells and are known to act as oncogenes in some haematopoietic malignancies (Eklund, 2007).
Repeated duplication events have given rise to 39 HOX genes in mammals, divided into four groups (A–D) in tightly linked clusters on different chromosomes (Hoegg and Meyer, 2005). The HOX genes are also divided into paralogue groups – genes that have the equivalent position in each cluster (1–13) – thus HOXA1, for example, is the 3′ most gene in cluster A (Scott, 1993). Whereas some HOX genes have distinct functions in specific contexts, many others have overlapping or redundant functions, both during early development (McIntyre et al, 2007) and in malignant cells (Eklund, 2007). This redundancy in HOX function is based in part upon the binding of similar DNA sequences and also on the interaction of HOX proteins with a common set of co-factors including PBX and MEIS. PBX and MEIS modify the DNA-binding specificity of HOX proteins, influence the regulation of transcription and are required for many aspects of HOX function (Moens and Selleri, 2006).
In addition to haematological malignancies, the HOX genes are also expressed at high levels in many other solid malignancies, although with the exception of melanoma (Morgan et al, 2007), they are not known to have an oncogenic function. A number of studies, focusing on small groups of HOX genes, have shown that some are upregulated in lung cancer (Abe et al, 2006), but the functional significance of this is unknown. Here we present a comprehensive analysis of HOX expression in non-small-cell lung cancer (NSCLC) that reveals that many of the HOX genes have strongly elevated expression in malignant cells. Further, we show that antagonising HOX/PBX binding in the NSCLC cell lines A549 (Lieber et al, 1976) and H23 (Little et al, 1983) induces apoptosis in vitro and causes significant tumour shrinkage in A549 nude mouse models.
Materials and methods
Maintenance of A549 and H23 in culture
The NSCLC cell lines A549 and H23 were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). The cells were cultured in F-12K Nutrient Mixture Kaighn's Modification medium (Invitrogen, Paisley, UK) supplemented with 10% foetal calf serum (Invitrogen) and 1% penicillin (10 000 U ml−1)/streptomycin (10 mg ml−1) (Sigma, Gillingham, UK). Cell cultures were maintained at 37°C in a humidified, 5% CO2 incubator.
Synthesis of HXR9 and CXR9 peptides
HXR9 is an 18-amino-acid peptide consisting of the previously identified hexapeptide sequence that can bind to PBX and nine C-terminal arginine residues (R9) that facilitate cell entry (Morgan et al, 2007). The N-terminal and C-terminal amino bonds are in the D-isomer conformation, which has earlier been shown to extend the half-life of the peptide to 12 h in human serum (Morgan et al, 2007). CXR9 is a control peptide that lacks a functional hexapeptide sequence and that includes the R9 sequence. All peptides were synthesised using conventional column-based chemistry and purified to at least 80% (Biosynthesis Inc., Lewisville, TX, USA).
Imaging of cell cultures
Cells were plated in six-well plates in 2 ml of medium and allowed to recover for at least 24 h. When approximately 60% confluent, the cells were treated with the active peptide HXR9 (60 μ M) or the control peptide CXR9 (60 μ M) for 3 h. For phase-contrast micrographs, the cells were washed twice with cold PBS and visualised using a Nikon Eclipse TS100 inverted microscope and images recorded using a Nikon camera and capture software (Jencons).
Analysis of cell death and apoptosis
Cells were treated with HXR9 or CXR9 as described above. Assessment of cell viability was performed using the MTS assay (Promega, Madison, WI, USA) according to the manufacturer's instructions. Cells were harvested by incubating in trypsin-EDTA (Sigma) at 37°C until detached and dissociated. Apoptotic cells were identified using flow cytometry (Beckman Coulter Epics XL Flow) and the Annexin-V-PE apoptosis detection kit (BD Pharmingen, Franklin Lakes, NJ, USA) as described by the manufacturer's protocol.
RNA purification and reverse transcription
Total RNA was isolated from cells using the RNeasy Plus Mini Kit (Qiagen, Crawley, UK) by following the manufacturer's protocol. The RNA was denatured by heating to 65°C for 5 min. cDNA was synthesised from RNA using the Cloned AMV First Strand Synthesis Kit (Invitrogen) according to the manufacturer's instructions.
Quantitative PCR
Quantitative PCR was performed using the Stratagene MX3005P real-time PCR machine and the Brilliant SYBR Green QPCR Master Mix (Stratagene, La Jolla, CA, USA). Oligonucleotide primers were designed to facilitate the unique amplification of β-actin, c-Fos and each HOX gene.
Transcriptional profiling
Total RNA was extracted from A549 cells treated with CXR9 or HXR9 (60 μ M) for 3 h, and was used as a template to generate Cy3-labelled cRNA using the Low RNA Input Linear Amplification Kit (Agilent, Santa Clara, CA, USA). Each Cy3-cRNA was used as a probe on the Whole Human Genome Microarray (4 × 44K) slide. This microarray consists of 60-mer oligonucleotides with sequences representing more than 41 000 human genes. The microarray slides were scanned and data were extracted using the Agilent Feature Extraction Software (version 9.5.3). Data were analysed using GeneSpring GX software.
Mice and in vivo trial
All animal experiments were conducted in accordance with the United Kingdom Co-ordinating Committee on Cancer Research guidelines for the Welfare of Animals in Experimental Neoplasia (Workman et al, 1998) and approved by the St George's Hospital Medical School Ethical Review Committee. The mice were kept in positive pressure isolators in 12 h light/dark cycles and food and water were made available ad libitum.
Athymic nude mice were inoculated subcutaneously with a suspension of 2.5 × 106 A549 cells in culture media (100 μl). Once tumours reached volumes of approximately 100 mm3, mice received an initial dose of 100 mg kg−1 CXR9 or HXR9 either intraperitoneally or intratumorally, with subsequent dosing of 10 mg kg−1 twice weekly. Each group contained 10 mice. The mice were monitored carefully for signs of distress, including behavioural changes and weight loss. Animals were euthanised if the tumour volume exceeded 400 mm3.
Statistical analysis of the data
Data are given as means±s.e.m. of multiple independent experiments. Significant effects were determined using Student's t-test.
Results
HOX gene expression in NSCLC
The HOX proteins of paralogue groups 1–9 bind the co-factor PBX and are thus potential targets of HXR9. To examine the expression of HOX genes in these paralogue groups, RNA was extracted from three separate cultures of A549 and H23 together with six primary NSCLC tumours retrieved during biopsy and corresponding normal adjacent tissue. Quantitative PCR was used to assess the relative numbers of HOX transcripts. Differences in transcription are apparent for many of the HOX genes (Figure 1). Notably, a number of genes in the HOXA group are strongly expressed in normal tissue, but are downregulated in both primary tumours and A549, including HOXA3 and HOXA5. Conversely, many members of the HOXC and HOXD groups are strongly upregulated in the primary tumours and A549, including HOXC4, HOXC8, HOXC9, HOXC13, HOXD8 and HOXD10. H23 cells also express HOX genes but to a considerably lesser degree than the other cell types studied.

HOX gene expression in primary lung NSCLC tumours, normal adjacent tissue (NAT) and derived cell lines. The expression of each HOX gene was determined by Q-PCR and is presented as a ratio to the amount of β-actin transcript ( × 1000). Error bars show the s.e.m.
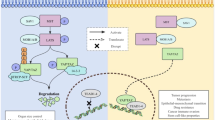
Disrupting the interaction between HOX and PBX triggers apoptosis in NSCLC cells
The high levels of HOX expression in NSCLC together with the potential oncogenic function of HOX genes (Eklund, 2007) suggest that they may be a therapeutic target in this disease. However, the high level of functional redundancy exhibited by the HOX transcription factors, together with our observation that many different HOX genes are expressed, makes a conventional genetic knockdown approach difficult to use. For this reason, we chose to target the interaction between HOX proteins and the PBX co-factor. HOX/PBX dimers have an enhanced binding affinity and specificity for DNA and are able to activate or repress the transcription of a subset of target genes (Phelan et al, 1994; Piper et al, 1999; Morgan et al, 2000). Dimerisation is mediated by a six-amino-acid region found in the majority of HOX proteins (the ‘hexapeptide’ motif (Piper et al, 1999)). A peptide mimic of this region linked to a polyarginine sequence (that facilitates cell entry (Jiang et al, 2004)) might act as a competitive inhibitor of HOX/PBX binding. We therefore synthesised a peptide that consists of the hexapeptide sequence linked to nine arginine residues (HXR9) and a control peptide lacking a functional hexapeptide sequence (CXR9). Using a fluorescently tagged version of HXR9, we demonstrated that the peptide was taken up by NSCLC cells in culture and became concentrated in the nucleus within 2 h of application (Figure 2A).

HXR9 blocks HOX/PBX dimer formation in NSCLC cells. (A) HXR9 uptake by A549 cells. A fluorescent micrograph of A549 cells incubated with 15 μ M HXR9-FITC (green) for 2 h, combined with the corresponding phase-contrast image. HXR9 is primarily located in the nucleus. (B) HOXA2 expression is maintained in part through a HOX/PBX-binding site in its promoter. Correspondingly, Q-PCR analysis of H23 cells treated with 60 μ M HXR9 for 2 h reveals that HOXA2 expression is significantly reduced upon HXR9 treatment. (C) ChIP analysis of the HOXA2 promoter. Chromatin was extracted from HXR9- or CXR9-treated cells. After cleavage, fragments were immunoprecipitated using an anti-HOXA2 or an anti-PBX antibody, and the number of copies of the HOXA2 promoter region recovered was determined by Q-PCR. A reduced number of HOXA2 promoter sequences is recovered from HXR9-treated cells when immunoprecipitating with anti-HOXA2 or anti-PBX antibodies, whereas no significant difference is apparent when equal amounts of chromatin are used without selection.
To determine whether HXR9 can specifically disrupt HOX/PBX formation (and consequently DNA binding), we studied the effects of HXR9 on HOXA2 expression. HOXA2, like a number of other HOX genes, is positively regulated by binding of HOX/PBX proteins to consensus sequences in the HOXA2 promoter (Tümpel et al, 2007). Hence, if HXR9 can disrupt this interaction, it should reduce HOXA2 expression, which it does (Figure 2B). To show that HXR9 actually prevents the interaction between HOX and PBX proteins we used chromatin immune precipitation analysis of HXR9- and CXR9-treated H23 cells. The presence of the HOX/PBX-binding site in the HOXA2 promoter results in the enrichment of this sequence when chromatin is selected using either an anti-HOXA2 or an anti-PBX2 antibody. However, pre-treatment of cells with HXR9, but not CXR9, causes a significant reduction in the number of HOXA2 promoter copies recovered using this technique (Figure 2C), indicating that HXR9 does indeed directly disrupt the formation of a HOX/PBX complex.
Given the high level of HOX expression in primary tumours and A549 and H23 cells, we investigated whether HXR9 could trigger cell death in a similar manner to that observed for melanoma cells (Morgan et al, 2007). Treatment of cultures with HXR9 (60 μ M) for 3 h resulted in a high degree of cell death whereas CXR9 had no discernable effects. The IC50 (24 h) for HXR9 was determined to be 32.5 μ M (s.e.m. 0.6 μ M) in A549 and 69 μ M (s.e.m. 1.2 μ M) in H23, whereas that for CXR9 was in excess of 200 μ M in both cell lines (data not shown).
To distinguish between apoptotic and necrotic death, cells were analysed using flow cytometric analysis after labelling with the vital dye 7-Amino-actinomycin D (7-AAD) or Annexin-V-FITC. 7-Amino-actinomycin D can enter only dead cells, whereas Annexin-V-FITC recognises changes in the plasma membrane that are characteristic of apoptosis (Koopman et al, 1994). A substantial number of A549 cells treated with HXR9 (60 μ M) were shown to be undergoing some form of cell death (Figure 3A and B). When compared with untreated or CXR9-treated cells, a significant proportion of HXR9-treated cells (P<0.005) were in either early or late apoptosis, or were necrotic (P<0.05). Similar changes were also observed for H23 cells (Figure 3C).

HXR9 triggers apoptosis in A549 and H23 NSCLC cells. Flow cytometric analysis of Annexin-V-PE-stained A549 and H23 cells demonstrated induction of apoptosis in cultures treated with HXR9. Data are the mean±s.e.m. of four independent experiments. *P<0.05, significantly different from CXR9-treated cells; **P<0.005, highly significantly different from CXR9-treated cells; ***P<0.0001, extremely significantly different from CXR9-treated cells. (A) Example plots of flow cytometric data for untreated and HXR9-treated A549 cells. x axis – Annexin-V staining indicating degree of apoptotic changes in the cell membrane; y axis – 7-AAD vital dye counterstaining as a measure of cell permeability. (B) Summary of flow cytometry data for A549 cells and (C) for H23 cells. Error bars represent the s.d. (n=4); ‘V’ – viable cells not in apoptosis, ‘EA’ – cells in early apoptosis, ‘LA’ – cells in late apoptosis and ‘N’ – necrotic cells.
HXR9 causes transcriptional changes
To study the transcriptional changes that occur when HOX/PBX dimers are disrupted by HXR9 we profiled the transcriptome of CXR9- and HXR9-treated cells using a whole human genome microarray. RNA was extracted from A549 cells treated with 60 μ M HXR9 or CXR9 for 3 h and used as a template to generate Cy3-labelled cRNA for microarray screening. This revealed that a number of genes were highly upregulated, including a number that had earlier been observed in melanoma cells (Morgan et al, 2007). Most significant among these were vFos and FosB, both of which were upregulated more than five-fold in HXR9-treated cells compared with controls (Figure 4A). The relative magnitude of these changes was confirmed using semi-quantitative PCR (Figure 4B).

HXR9 causes transcriptional changes. (A) Subsequent to treatment with CXR9 or HXR9, RNA was extracted from A549 cells and analysed by microarray. There was no significant change in the transcription of the majority of genes, although five of them showed an increase, and eight a decrease, of five-fold or greater. (B) Semi-quantitative PCR was used to confirm the microarray findings. Gene expression was determined relative to β-actin expression and results are presented as a ratio with the expression of each gene in untreated cells. Error bars represent the s.e. of the mean.
HXR9 blocks A549 tumour growth in vivo
To establish whether HXR9 could block A549 tumour growth in vivo, A549 flank tumours were established in nude mice. When these had reached an average volume of 100 mm3, twice weekly treatments of HXR9 or CXR9 were administered by injection directly either into the tumour or into the peritoneum. After 18 days of treatment, the tumours of HXR9-treated mice were considerably smaller than those of the control groups (Figure 5A), and histological analysis revealed changes in tumour architecture consistent with widespread cell death (Figure 5B).

HXR9 blocks tumour growth in vivo. (A) Thymic nude mice were inoculated subcutaneously with 2.5 × 106 A549 cells. When tumours reached volumes of approximately 100 mm3, the animals received an initial dose of 100 mg kg−1 of CXR9 or HXR9 either in to the peritoneum or intratumorally, with subsequent dosing of 10 mg kg−1 twice weekly. (B) Structural changes in HXR9-treated tumours. Tumours were excised from mice after the 18-day treatment cycle (twice weekly intraperitoneal administration of 10 mg kg−1 HXR9), embedded and sectioned. Distinct differences in tumour architecture are apparent in HXR9-treated mice, most notably in areas where extensive cell death has occurred (arrow heads). Scale bar: 100 μm.
Discussion
The HOX genes are well established as having oncogenic potential in a range of haematological malignancies (Eklund, 2007), and recent studies have indicated that they also allow cell survival in melanoma by blocking the transcription of pro-apoptotic genes (Morgan et al, 2007). Here we have shown that disrupting the interaction between HOX and PBX proteins is sufficient to cause apoptosis in the NSCLC-derived cell lines, A549 and H23. Although earlier studies have also shown HOX genes to be dysregulated in NSCLC (Abe et al, 2006), to our knowledge this is the first report to describe these changes in a comprehensive manner allowing a direct comparison between expression in normal lung tissue, primary tumours and derived cell lines. This reveals that there is a large increase in transcription in most HOX genes of the HOXC and HOXD families when comparing normal lung tumour with primary tumours, and furthermore, this change is also present in A549 and H23, indicating that there has been a stable change in the transcriptome.
Analysis of changes in transcription upon the addition of HXR9 revealed that the Early Growth Response 1 (EGR1) gene is upregulated by more than 20-fold. EGR1 has been shown to mediate apoptosis in a number of malignancies (Pignatelli et al, 2003; Yu et al, 2007), possibly by activating the transcription of pro-apoptotic genes such as PTEN, and EGR1 can also block angiogenesis and tumour invasion (Lucerna et al, 2006; Yu et al, 2007). Further, high levels of EGR1 correlate with improved survival in NSCLC patients (Ferraro et al, 2005) and is upregulated when NSCLC cells are treated with the apoptosis-inducing retinoid 6-[3-(1-adamantyl)-4-hydroxyphenyl]-2-naphthalene carboxylic acid (Sakaue et al, 2001).
It is also noteworthy that HXR9-treated A549 cells show a large increase in the transcription of vFos, cFos and FosB. A similar response is apparent in melanoma cells in which a large increase in cFos transcription mediates the pro-apoptotic effect of HXR9 (Morgan et al, 2007). cFos and its binding partner Jun are both members of the bZIP superfamily of transcription factors, which are characterised by a basic DNA-binding domain combined with a leucine zipper region (Hess et al, 2004). JUN can homodimerise to form the AP-1 transcription factor, which activates the expression of a number of genes involved in cell cycle regulation, including cyclin D1 (Albanese et al, 1995). However, an elevated cFos expression can also lead to apoptosis in hepatoma cells (Kalra and Kumar, 2004) and hepatocytes (Mikula et al, 2003). Furthermore, recent studies have shown that cFos transcriptionally represses the key antiapoptotic gene c-FLIP(L), greatly sensitising prostate cancer cells to TRAIL-induced apoptosis (Li et al, 2007; Zhang et al, 2007). Hence strategies to increase cFos transcription could be valuable in making malignant cells vulnerable to apoptosis, and HXR9 may synergise with TRAIL and other drugs that induce apoptosis to facilitate enhanced cell killing across a broad range of tumour types.
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Abe M, Hamada J, Takahashi O, Takahashi Y, Tada M, Miyamoto M, Morikawa T, Kondo S, Moriuchi T (2006) Disordered expression of HOX genes in human non-small cell lung cancer. Oncol Rep 15: 797–802
Abramovich C, Humphries RK (2005) Hox regulation of normal and leukemic hematopoietic stem cells. Curr Opin Hematol 12: 210–216
Albanese C, Johnson J, Watanabe G (1995) Transforming p21ras mutants and c-Ets-2 activate the cyclin D1 promoter through distinguishable regions. J Biol Chem 270: 23589–23597
Eklund EA (2007) The role of HOX genes in malignant myeloid disease. Curr Opin Hematol 14: 85–89
Ferraro B, Bepler G, Sharma S, Cantor A, Haura EB (2005) EGR1 predicts PTEN and survival in patients with non-small-cell lung cancer. J Clin Oncol 23: 1921–1926
Hess J, Angel P, Schorpp-Kistner M (2004) AP-1 subunits: quarrel and harmony among siblings. J Cell Sci 11: 5965–5973
Hoegg S, Meyer A (2005) Hox clusters as models for vertebrate genome evolution. Trends Genet 21: 421–424
Iimura T, Pourquié O (2007) Hox genes in time and space during vertebrate body formation. Dev Growth Differ 49: 265–275
Jiang T, Olson ES, Nguyen QT, Roy M, Jennings PA, Tsien RY (2004) Tumor imaging by means of proteolytic activation of cell-penetrating peptides. Proc Natl Acad Sci USA 101: 17867–17872
Kalra N, Kumar V (2004) c-Fos is a mediator of the c-myc-induced apoptotic signaling in serum-deprived hepatoma cells via the p38 mitogen-activated protein kinase pathway. J Biol Chem 279: 25313–25319
Koopman G, Reutelingsperger CP, Kuijten GA, Keehnen RM, Pals ST, van Oers MH (1994) Annexin V for flow cytometric detection of phosphatidylserine expression on B cells undergoing apoptosis. Blood 84: 1415–1420
Lieber M, Smith B, Szakal A, Nelson-Rees W, Todaro G (1976) A continuous tumor-cell line from a human lung carcinoma with properties of type II alveolar epithelial cells. Int J Cancer 17: 62–70
Li W, Zhang X, Olumi AF (2007) MG-132 sensitizes TRAIL-resistant prostate cancer cells by activating c-Fos/c-Jun heterodimers and repressing c-FLIP(L). Cancer Res 67: 2247–2255
Little CD, Nau MM, Carney DN, Gazdar AF, Minna JD (1983) Amplification and expression of the c-myc oncogene in human lung cancer cell lines. Nature 306: 194–196
Lucerna M, Pomyje J, Mechtcheriakova D, Kadl A, Gruber F, Bilban M, Sobanov Y, Schabbauer G, Breuss J, Wagner O, Bischoff M, Clauss M, Binder BR, Hofer E (2006) Sustained expression of early growth response protein-1 blocks angiogenesis and tumor growth. Cancer Res 66: 6708–6713
McIntyre DC, Rakshit S, Yallowitz AR, Loken L, Jeannotte L, Capecchi MR, Wellik DM (2007) Hox patterning of the vertebrate rib cage. Development 134: 2981–2989
Mikula M, Gotzmann J, Fischer AN (2003) The proto-oncoprotein c-Fos negatively regulates hepatocellular tumorigenesis. Oncogene 22: 6725–6738
Moens CB, Selleri L (2006) Hox cofactors in vertebrate development. Dev Biol 291: 193–206
Morgan R, In der Rieden P, Hooiveld MH, Durston AJ (2000) Identifying HOX paralog groups by the PBX-binding region. Trends Genet 16: 66–67
Morgan R, Pirard P, Shears S, Sohal S, Pettengell R, Pandha HS (2007) Antagonism of HOX/PBX dimer formation blocks the in vivo proliferation of melanoma. Cancer Res 67: 5806–5813
Phelan ML, Sadoul R, Featherstone MS (1994) Functional differences between HOX proteins conferred by two residues in the homeodomain N-terminal arm. Mol Cell Biol 14: 5066–5075
Pignatelli M, Luna-Medina R, Pérez-Rendón A, Santos A, Perez-Castillo A (2003) The transcription factor early growth response factor-1 (EGR-1) promotes apoptosis of neuroblastoma cells. Biochem J 373: 739–746
Piper DE, Batchelor AH, Chang CP, Cleary ML, Wolberger C (1999) Structure of a HoxB1-Pbx1 heterodimer bound to DNA: role of the hexapeptide and a fourth homeodomain helix in complex formation. Cell 96: 587–597
Sakaue M, Adachi H, Dawson M, Jetten AM (2001) Induction of Egr-1 expression by the retinoid AHPN in human lung carcinoma cells is dependent on activated ERK1/2. Cell Death Differ 8: 411–424
Scott MP (1993) A rational nomenclature for vertebrate homeobox (HOX) genes. Nucleic Acids Res 21: 1687–1688
Tümpel S, Cambronero F, Ferretti E, Blasi F, Wiedemann LM, Krumlauf R (2007) Expression of Hoxa2 in rhombomere 4 is regulated by a conserved cross-regulatory mechanism dependent upon Hoxb1. Dev Biol 302: 646–660
Workman P, Twentyman P, Balkwill F, Balmain A, Chaplin D, Double J, Embleton J, Newell D, Raymond R, Stables J, Stephens T, Wallace J (1998) United Kingdom Co-ordinating Committee on Cancer Research (UKCCCR) guidelines for the welfare of animals in experimental neoplasia (second edition). Br J Cancer 77: 1–10
Yu J, Baron V, Mercola D, Mustelin T, Adamson ED (2007) A network of p73, p53 and Egr1 is required for efficient apoptosis in tumor cells. Cell Death Differ 14: 436–446
Zhang X, Zhang L, Yang H, Huang X, Otu H, Libermann TA, Dewolf WC, Khosravi-Far R, Olumi AF (2007) c-Fos as a proapoptotic agent in TRAIL-induced apoptosis in prostate cancer cells. Cancer Res 67: 9425–9434
Acknowledgements
This study was supported by grants from Cancer Research-UK [(7822/A383)] and the Prostate Project.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Plowright, L., Harrington, K., Pandha, H. et al. HOX transcription factors are potential therapeutic targets in non-small-cell lung cancer (targeting HOX genes in lung cancer). Br J Cancer 100, 470–475 (2009). https://doi.org/10.1038/sj.bjc.6604857
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6604857
Keywords
This article is cited by
-
Peptides as multifunctional players in cancer therapy
Experimental & Molecular Medicine (2023)
-
Loss of DLX3 tumor suppressive function promotes progression of SCC through EGFR–ERBB2 pathway
Oncogene (2021)
-
Long non-coding RNA HOTAIR and HOTTIP as potential biomarkers for hepatitis C virus genotype 4-induced hepatocellular carcinoma
Egyptian Journal of Medical Human Genetics (2020)
-
Mapping the native interaction surfaces of PREP1 with PBX1 by cross-linking mass-spectrometry and mutagenesis
Scientific Reports (2020)
-
Clinical significance of long non-coding RNA HOTTIP in early-stage non-small-cell lung cancer
BMC Pulmonary Medicine (2019)
