
Abstract
Retrospective analysis has shown that activating mutations in exons 18–21 of the epidermal growth factor receptor (EGFR) gene are a predictor of response to gefitinib. We conducted a phase II trial to evaluate the efficacy and safety of gefitinib as first-line therapy for advanced non-small cell lung cancer (NSCLC) with EGFR mutations. Patients with stage IIIB or IV chemotherapy-naïve NSCLC with EGFR mutation were treated with 250 mg gefitinib daily. For mutational analysis, DNA was extracted from paraffin-embedded tissues and EGFR mutations were analysed by direct sequence of PCR products. Twenty (24%) of the 82 patients analysed had EGFR mutations (deletions in or near E746-A750, n=16; L858R, n=4). Sixteen patients were enrolled and treated with gefitinib. Twelve patients had objective response and response rate was 75% (95% CI, 48–93%). After a median follow-up of 12.7 months (range, 3.1–16.8 months), 10 patients demonstrated disease progression, with median progression-free survival of 8.9 months (95% CI, 6.7–11.1 months). The median overall survival time has not yet been reached. Most of the toxicities were mild. This study showed that gefitinib is very active and well tolerated as first-line therapy for advanced NSCLC with EGFR mutations.
Similar content being viewed by others


Main
Lung cancer has long been the leading cause of cancer death in North America and became the leading cause of cancer death in Japan in 1998 (Kuroishi et al, 1999). Both platinum-based and taxane-based chemotherapy offer modest efficacy and survival advantages over best supportive care (BSC) alone for chemotherapy-naïve patients with advanced non-small cell lung cancer (NSCLC). Nonetheless, objective response rate (RR) is at most 30–40%, and median survival time (MST) is only 8–10 months owing to frequent recurrence and metastasis (Non-Small Cell Lung Cancer Collaborative Group, 1995; Schiller et al, 2002). Other promising drug therapy has therefore long been awaited.
The epidermal growth factor receptor (EGFR) is a 170 kDa tyrosine kinase (TK) that dimerises and phosphorylates several tyrosine residues upon binding of several specific ligands. These phosphorylated tyrosines serve as the binding sites for several signal transducers that initiate multiple signalling pathways which lead to cell proliferation, differentiation, migration and metastasis, angiogenesis and antiapoptosis (Arteaga, 2002). Because EGFR is highly expressed in 43–89% of NSCLC (Scagliotti et al, 2004), EGFR-tyrosine kinase inhibitors (EGFR-TKIs) such as gefitinib and erlotinib have emerged as particularly promising target drugs for treating NSCLC.
Two phase II trials for patients previously treated with chemotherapy, the Iressa Dose Evaluation in Advanced Lung Cancer (IDEAL)-1 and -2, revealed favourable objective RRs of 10–20% and a disease control rate of 50% (Fukuoka et al, 2003; Kris et al, 2003). Subsequent randomised phase III trials were conducted; the Iressa NSCLC Trial Assessing Combination Treatment (INTACT)-1 and -2, which were randomised, placebo-controlled trials of cisplatin/gemcitabine or carboplatin/paclitaxel with or without gefitinib for chemotherapy-naïve patients. However, these did not indicate any additional benefit of gefitinib over conventional cytotoxic chemotherapy, as measured by RR and median and 1-year survival (Giaccone et al, 2004; Herbst et al, 2004). Moreover, another phase III trial, the Iressa Survival Evaluation in Lung Cancer (ISEL), showed no survival benefit of gefitinib over BSC as a salvage regimen (Thatcher et al, 2005). On balance therefore, the usefulness of gefitinib for advanced NSCLC remains controversial. Similar results were observed regarding another EGFR-TKI, erlotinib (Gatzemeier et al, 2004; Herbst et al, 2005), with the exception of the BR21 trial which showed survival benefit over BSC for erlotinib as salvage therapy (Shepherd et al, 2005).
Nevertheless, subgroups of patients, such as women, nonsmokers, patients with adenocarcinomas, and East Asian patients, tend to have higher objective responses and sometimes exhibit dramatic tumour shrinkage in response to these agents. It would therefore appear very important to discover biomarkers that predict gefitinib sensitivity, although EGFR protein expression, the most plausible candidate marker, does not correlate with gefitinib efficacy (Cappuzzo et al, 2003).
Recently, activating mutations in the TK domain of EGFR were reported to be strongly associated with clinical responsiveness to EGFR-TKIs (Lynch et al, 2004; Paez et al, 2004; Pao et al, 2004). Although these results are thought-provoking, no prospective trials have been reported to date regarding gefitinib monotherapy for advanced NSCLC with EGFR mutations. Here, we conducted a phase II trial to evaluate the efficacy and safety of gefitinib as first-line therapy for advanced NSCLC with EGFR mutations.
Materials and methods
Study design
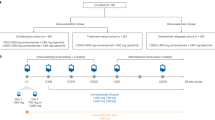
This single-arm, phase II clinical trial recruited patients at nine institutes in north-eastern Japan. This trial consisted of two stages. First, EGFR mutations in exons 18–21 of the accrued patients were analysed as described below. In the second stage, only those who had EGFR mutations were enrolled and treated with gefitinib. The primary end point was the objective RR to an intervention of gefitinib administration (250 mg daily). Secondary end points were toxicity and survival.
Patient eligibility
Eligible patients had histologically or cytologically confirmed stage IIIB or IV, chemotherapy-naïve NSCLCs with EGFR mutations. Recurrences after surgical resection were also eligible. Other eligibility criteria included: (a) age 20 years or older; (b) Eastern Cooperative Oncology Group (ECOG) performance status (PS) of 0–2; (c) measurable lesions; (d) adequate organ function (i.e., leucocyte count ⩾4000 mm−3, haemoglobin ⩾9.5 g dl−1, platelets ⩾100 000 mm−3, total bilirubin ⩽1.5 mg dl −1, AST and ALT ⩽2 times the upper limit of the reference range, serum creatinine ⩽1.5 mg/dl −1, PaO2 ⩾70 torr); and (e) life expectancy of 12 weeks or longer). Exclusion criteria comprised: (a) unstable angina, acute myocardial infarction or heart failure within the previous 3 months; (b) uncontrolled diabetes mellitus or hypertension; (c) active infection; (d) interstitial pneumonia or pulmonary fibrosis as determined from chest computed tomography (CT); (e) uncontrolled pleural effusion; (f) active gastrointestinal ulcer; (g) active metachronous cancer; (h) past history of severe hypersensitivity; (i) severe superior vena cava syndrome; and (j) pregnancy or breast-feeding. All patients were required to provide written informed consent. Trial protocol approval was obtained from the ethics committee or institutional review board (IRB) at each trial institute.
Treatment plan
Patients received 250 mg of gefitinib orally per day. In the event of unacceptable toxicity (defined as grade 3 or more) or deterioration of PS to 3 or 4, gefitinib was ceased until this toxicity resolved and/or PS improved to ⩽grade 2 within 3 weeks. If this did not occur, treatment was terminated. In the event of grade 2 or higher interstitial lung disease (ILD), treatment was also terminated. Dose reduction was not performed. Treatment was continued unless any of the following occurred: progressive disease (PD), unacceptable toxicity (as mentioned above), the study physician decided to terminate therapy or the patient withdrew consent. No systematic anticancer treatment, radiotherapy or pleurodesis was permitted during the trial. Salvage regimens were not restricted for patients with PD or those leaving the protocol.
Evaluation of efficacy and toxicity
Pretreatment evaluations consisted of the following: complete medical history, determination of PS, physical examination, haematologic and biochemical profiles, arterial blood gas examination, ECG, spirometry, chest X-ray, bone scan, CT of the chest, ultrasound or CT of the abdomen, and magnetic resonance imaging or CT of the whole brain. Evaluations performed during treatment included a weekly chest X-ray, biochemistry, complete blood count (including platelet and differential leucocyte counts), physical examination, determination of PS, and toxicity assessment. Moreover, for the early detection of ILD, spirometry and helical CT scan of the chest were performed once every 2 weeks for the initial 4 weeks. Imaging studies were scheduled every month to assess objective response.
Response evaluation criteria in solid tumours (RECIST) guidelines were used for evaluation of antitumour activity (Therasse et al, 2000). Complete response (CR) was defined as the complete disappearance of all clinically detectable tumours for at least 4 weeks. Partial response (PR) was defined as a ⩾30% decrease in the sum of the longest diameters of the target lesions for a minimum of 4 weeks with no new area of malignant disease. Progressive disease indicated at least a 20% increase in the sum of the longest diameter of the target lesions or a new malignant lesion. Stable disease (SD) was defined as insufficient shrinkage to qualify for PR and insufficient increase to qualify for PD. The minimum interval to qualify for SD was defined as 8 weeks. Responses were evaluated by the physician in charge and confirmed by independent reviewers at an extramural conference. Toxicity was graded according to the National Cancer Institute Common Toxicity Criteria (NCI-CTC) version 2.0. (National Cancer Institute, 1999).
Mutational analysis of the epidermal growth factor receptor gene
Tumour specimens were obtained during diagnostic or surgical procedures. For patients with recurrences after surgical resection, mutation status was analysed in specimens of the original primary sites. Biopsied or surgically resected specimens were fixed with formalin and embedded in paraffin. Whole paraffin-embedded tissue blocks or ⩾4 slices (5-μm thick) from blocks which were confirmed by each institute's pathologist to contain adequate malignant tumour were sent to First Department of Medicine, Hokkaido University. Genomic DNA was isolated from specimens using a DNeasy Tissue kit (Qiagen, Valencia, CA, USA) according to the manufacturer's instructions. For mutational analysis of the kinase domain of the EGFR, exons 18–21 were amplified with four pairs of primers as described previously (Paez et al, 2004), using a HotStarTaq DNA polymerase kit (Qiagen). Polymerase chain reaction products were purified with a PCR purification kit (Qiagen) and sequenced directly with an Applied Biosystems BigDye Terminator kit v3.1 (Applied Biosystems, Foster City, CA, USA) with an ABI PRISM 310 Genetic Analyzer. Both the forward and reverse sequences were analysed by BLAST, and chromatograms were manually reviewed. If the obtained sequences included mutation sequences, PCR amplification and sequencing analysis were repeated to confirm the results. Only the following mutations described in previous reports (Lynch et al, 2004; Paez et al, 2004; Pao et al, 2004) were regarded as mutation positive in the present trial; G719X in exon18, deletions in or near E746-A750 in exon 19, and L858R and L861Q in exon 21.
Statistical analysis
Simon's two-stage minimax design was used to determine the sample size and decision criteria for this phase II trial (Simon, 1989). With a target activity level of 70% (P1) and minimum RR of interest set at 30% (P0), we needed 14 evaluable patients to accept the hypothesis and a 5% significance level to reject it with 90% power. Assuming an inevaluability rate of ⩽15%, we projected an accrual of 16 patients. Progression-free survival (PFS) was defined as the interval between enrolment in this trial and the date of documented disease progression or death from any cause. Overall survival (OS) was defined as the interval between enrolment in this trial and death from any cause. If a patient was lost to follow-up, that patient was censored at the last date of contact. Median overall and progression-PFS were estimated by the Kaplan–Meier analysis method (Kaplan and Meier, 1958). All patients who were enrolled and treated with gefitinib were included in both efficacy and toxicity analyses. Data were updated as of 15 June 2006.
Results
Characteristics of patients undergoing epidermal growth factor receptor mutation analysis
From November 2004 to January 2006, 82 patients underwent analysis of EGFR mutation status. Patient characteristics are listed in Table 1. Forty-nine patients (60%) were female. Median age was 65 years (range, 36–83 years). The most common tumour histological type was adenocarcinoma in 72 patients (87%). Thirty-eight patients (46%) were never-smokers. Tissue samples from 44 patients (54%) were obtained by transbronchial biopsy.
Comparison of epidermal growth factor receptor mutation status and clinicopathological characteristics
Twenty patients (24%) had EGFR mutations (deletions in or near E746-A750, n=16; L858R, n=4). Epidermal growth factor receptor mutation status and patient clinicopathological characteristics are compared in Table 2. Although no significant differences were apparent between mutation status and age, sex, histology or procedure used to obtain tumour specimens, EGFR mutations were more frequently observed in never-smokers than in smokers (39 vs 11%, P<0.01).
Response to gefitinib in patients with epidermal growth factor receptor mutations
Of 20 patients who had EGFR mutations, four were not enrolled to the subsequent phase II trial. One patient withdrew his consent before enrolment, one had no appropriate measurable lesions, and two were misclassified when staging. We therefore excluded these four patients before enrolment and assembled no more data from them. Excluding these four patients, 16 patients (median age, 68; male/female, 3/13; adenocarcinoma/squamous cell carcinoma, 15/1; current/former/never smokers, 2/1/13) were enrolled in the phase II trial and treated with gefitinib. Details of EGFR mutations and clinicopathologic features in patients receiving gefitinib are described in Table 3. Of the 16 enrolled patients, 12 achieved objective responses (two CR and 10 PR) with an overall RR of 75% (95% CI, 48–93%), one (6%) had SD and three (19%) had PD as the best response. Disease control rate (CR+PR+SD) was 81% (95% CI, 54–96%). Although the number of L858R patients was small (n=3), no significant difference was evident between type of mutation and RR (exon 19 deletions, 83 vs L858R, 67%; P=0.87). To date, only two patients have died, all owing to disease progression. Of the remaining 14 patients who are still alive, six maintain PR. Hence, MST has not been reached (Figure 1), and we instead evaluated median PFS. At the time of this analysis, with a median follow-up time of 12.7 months, median PFS was 8.9 months (95% CI, 6.7–11.1 months).

(A) Overall survival and (B) PFS of all eligible patients (n=16) were calculated according to the Kaplan–Meier method. Median survival time has not yet been reached and median PFS was 8.9 months (95% CI, 6.7–11.1 months).
Safety and toxicity
Toxicity was evaluated in all eligible patients (Table 4). The most common manifestations of toxicity were dermatological. One patient experienced grade 3 rash and terminated gefitinib treatment on day 81; however, she achieved CR on day 29 and no recurrence was detected until day 228 without any second-line treatment for 147 days. Another frequently experienced adverse effect was hepatotoxicity (elevated AST/ALT). One patient required a long treatment interruption because of grade 3 hepatotoxicity and she discontinued the protocol on day 266. A further patient experienced grade 3 gastrointestinal ulcer and grade 4 anaemia and terminated gefitinib treatment on day 56 because of PD.
Interstitial lung disease is the most problematic toxicity in gefitinib treatment in Japan (Inoue et al, 2003). In the present trial, one patient experienced grade 1 ILD on day 30, leading to the termination of gefitinib treatment. This patient was asymptomatic; ILD was barely detectable on chest X-ray and was recognised only on chest CT, which revealed a patchy, ground-glass opacity with centrilobular distribution throughout the lung (Figure 2). This was assumed to be hypersensitivity-type ILD, and it improved without steroid administration.

Chest X-ray (A) and CT (B) on day 30 in the patient who developed ILD. Interstitial lung disease was hardly recognisable on chest X-ray, whereas chest CT revealed a patchy, ground-glass opacity with centrilobular distribution throughout the whole lung.
Second-line treatment after disease progression
Of 10 patients who became refractory to gefitinib owing to disease progression, six received second-line treatment. Two received carboplatin plus paclitaxel; one had PR and another had PD as second-line treatment. Another two patients received gemcitabine plus vinorelbine; one patient had PR and another had PD as second-line treatment. A further patient terminated gefitinib monotherapy because of grade 3 hepatotoxicity, recovered after 8 weeks treatment interruption and then resumed gefitinib. The other patient, in whom PD was diagnosed owing to development of a new bone metastasis, resumed gefitinib monotherapy after radiation therapy to the bone metastasis, as his primary and other lesions were still controlled by gefitinib. The latter two patients also remained alive at the most recent follow-up.
Discussion
As two separate groups reported somatic mutations of the EGFR TK domain in NSCLC in May 2004 (Lynch et al, 2004; Paez et al, 2004), over 15 studies on mutational analysis of this domain (exons 18–21) in over 3000 patients have been reported by different groups around the world (Chan et al, 2006). The collective data indicate an overall mutation rate of 17%, but this was higher in East Asians, never-smokers, women, and patients with adenocarcinoma. Notably, patients with mutated EGFR had a much higher RR to gefitinib than those with wild-type EGFR (77 vs 23%). Several groups also reported prolonged time to progression (range, 7.6–21.7 months) and OS time (range, 13.0–30.5 months) in patients with EGFR mutations (Chou et al, 2005; Cortes-Funes et al, 2005; Han et al, 2005; Kim et al, 2005; Mitsudomi et al, 2005; Takano et al, 2005; Taron et al, 2005; Zhang et al, 2005). However, retrospective analysis of the mutation status of patients enrolled in the previous large clinical trials of EGFR-TKI (Bell et al, 2005; Eberhard et al, 2005; Tsao et al, 2005) has not demonstrated a significant relationship between presence of EFGR mutation and response to such agents. Hence, to elucidate the efficacy of gefitinib in patients with EGFR mutations, the present prospective trial is warranted.
In this phase II trial, we demonstrated an extremely high objective RR of 75% (95% CI, 48–93%), median PFS of 8.9 months, and unattained OS, consistent with previous retrospective analyses. These results were much better than those for standard platinum-containing regimens as first-line therapy (Schiller et al, 2002). Therefore, although the real survival benefit needs to be examined in future randomised phase III trials, our results clearly demonstrated that gefitinib has considerable activity in patients with EGFR mutation, even as first-line therapy.
Despite such a good response to gefitinib, retrospective analysis of INTACT trials shows that patients with EGFR mutation also tend to be more sensitive to platinum-based chemotherapy than those with wild-type EGFR (Bell et al, 2005). This matter also needs to be addressed in future randomised phase III trials.
To date, two phase II clinical trials of gefitinib monotherapy as first-line therapy have been performed that did not consider EGFR mutation status. One evaluated gefitinib treatment without any patient selection (Niho et al, 2006) and the other was conducted with never-smokers (Lee et al, 2005). Although the former showed an unimpressive RR of 30%, the latter demonstrated high gefitinib efficacy; objective response, median PFS, and 1-year survival were 69%, 33 weeks and 73%, respectively. Although their observed responses were favourable and close to those observed in the present trial, patient selection based on smoking history might have the disadvantage of excluding the smokers with EGFR mutations who could have the same sensitivity to gefitinib as never-smokers.
Toxicity observed in the present trial was mostly favourable when compared to previous clinical trials of gefitinib (Fukuoka et al, 2003; Kris et al, 2003; Giaccone et al, 2004; Herbst et al, 2004; Thatcher et al, 2005) and of standard chemotherapeutic regimens (Schiller et al, 2002). Interstitial lung disease is the most problematic toxicity in gefitinib treatment in Japan where an incidence of 3.5% and a fatality rate of 1.6% have been reported (Inoue et al, 2003; Ando et al, 2006). Accordingly, in the present trial, we planned to conduct biweekly chest CT for early detection of ILD during the initial treatment period. Despite one case of ILD that occurred in the present trial, the initial biweekly CT detected asymptomatic grade 1 ILD better than has been reported previously. As ILD was detected at an early stage, it may have responded to discontinuation of gefitinib.
One problem related to patient selection based on mutation status is the method of detecting EGFR mutations. In the present trial, we used a direct sequence method with paraffin-embedded tissues. This method is conventional but is complex, expensive, and time consuming for daily clinical practice. Recently, simple, sensitive, and rapid detection methods such as PNA-LNA clump and LightCycler PCR assay have been developed (Nagai et al, 2005; Sasaki et al, 2005) and could resolve such problems. Clinical trials using these methods are being conducted by multiple groups.
In conclusion, gefitinib treatment as first-line therapy for advanced NSCLC with EGFR mutations demonstrated promising activity and a good toxicity profile. Randomised phase III trials comparing gefitinib and standard platinum-based chemotherapy for patients with EGFR mutations are now being conducted and have the potential to change our daily clinical practice with respect to advanced NSCLC.
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Ando M, Okamoto I, Yamamoto N, Takeda K, Tamura K, Seto T, Ariyoshi Y, Fukuoka M (2006) Predictive factors for interstitial lung disease, antitumor response, and survival in non-small-cell lung cancer patients treated with gefitinib. J Clin Oncol 24: 2549–2556
Arteaga CL (2002) Overview of epidermal growth factor receptor biology and its role as a therapeutic target in human neoplasia. Semin Oncol 29: 3–9
Bell DW, Lynch TJ, Haserlat SM, Harris PL, Okimoto RA, Brannigan BW, Sgroi DC, Muir B, Riemenschneider MJ, Iacona RB, Krebs AD, Johnson DH, Giaccone G, Herbst RS, Manegold C, Fukuoka M, Kris MG, Baselga J, Ochs JS, Haber DA (2005) Epidermal growth factor receptor mutations and gene amplification in non-small-cell lung cancer: molecular analysis of the IDEAL/INTACT gefitinib trials. J Clin Oncol 23: 8081–8092
Cappuzzo F, Gregorc V, Rossi E, Cancellieri A, Magrini E, Paties CT, Ceresoli G, Lombardo L, Bartolini S, Calandri C, de Rosa M, Villa E, Crino L (2003) Gefitinib in pretreated non-small cell lung cancer (NSCLC): analysis of efficacy and correlation with HER2 and epidermal growth factor receptor expression in locally advanced or metastatic NSCLC. J Clin Oncol 21: 2658–2663
Chan SK, Gulick WJ, Hill ME (2006) Mutations of the epidermal growth factor receptor in non-small cell lung cancer – Search and destroy. Eur J Cancer 42: 17–23
Chou TY, Chiu CH, Li LH, Hsiao CY, Tzen CY, Chang KT, Chen YM, Perng RP, Tsai SF, Tsai CM (2005) Mutation in the tyrosine kinase domain of epidermal growth factor receptor is a predictive and prognostic factor for gefitinib treatment in patients with non-small cell lung cancer. Clin Cancer Res 11: 3750–3757
Cortes-Funes H, Gomez C, Rosell R, Valero P, Garcia-Giron C, Velasco A, Izquierdo A, Diz P, Camps C, Castellanos D, Alberola V, Cardenal F, Gonzalez-Larriba JL, Vieitez JM, Maeztu I, Sanchez JJ, Queralt C, Mayo C, Mendez P, Moran T, Taron M (2005) Epidermal growth factor receptor activating mutations in Spanish gefitinib-treated non-small-cell lung cancer patients. Ann Oncol 16: 1081–1086
Eberhard DA, Johnson BE, Amler LC, Goddard AD, Heldens SL, Herbst RS, Ince WL, Janne PA, Januario T, Johnson DH, Klein P, Miller VA, Ostland MA, Ramies DA, Sebisanovic D, Stinson JA, Zhang YR, Seshagiri S, Hillan KJ (2005) Mutations in the epidermal growth factor receptor and in KRAS are predictive and prognostic indicators in patients with non-small-cell lung cancer treated with chemotherapy alone and in combination with erlotinib. J Clin Oncol 23: 5900–5909
Fukuoka M, Yano S, Giaccone G, Tamura T, Nakagawa K, Douillard JY, Nishiwaki Y, Vansteenkiste J, Kudoh S, Rischin D, Eek R, Horai T, Noda K, Takata I, Smit E, Averbuch S, Macleod A, Feyereislova A, Dong RP, Baselga J (2003) Multi-institutional randomized phase II trial of gefitinib for previously treated patients with advanced non-small cell lung cancer. J Clin Oncol 21: 2237–2246
Gatzemeier U, Pluzanska A, Szczesna A, Kaukel E, Roubec J, Brennscheidt U, De Rosa F, Mueller B, Von Pawel J, for the TALENT Study Investigators (2004) Results of a phase III trial of erlotinib (OSI-774) combined with cisplatin and gemcitabine (GC) chemotherapy in advanced non-small cell lung cancer (NSCLC). Proc Am Soc Clin Onc 22: 7010
Giaccone G, Herbst RS, Manegold C, Scagliotti G, Rosell R, Miller V, Natale RB, Schiller JH, Von Pawel J, Pluzanska A, Gatzemeier U, Grous J, Ochs JS, Averbuch SD, Wolf MK, Rennie P, Fandi A, Johnson DH (2004) Gefitinib in combination with gemcitabine and cisplatin in advanced non-small-lung cancer: a phase III trial-INTACT 1. J Clin Oncol 22: 777–784
Han SW, Kim TY, Hwang PG, Jeong S, Kim J, Choi IS, Oh DY, Kim JH, Kim DW, Chung DH, Im SA, Kim YT, Lee JS, Heo DS, Bang YJ, Kim NK (2005) Predictive and prognostic impact of epidermal growth factor receptor mutation in non-small-cell lung cancer patients treated with gefitinib. J Clin Oncol 23: 2493–2501
Herbst RS, Giaccone G, Schiller JH, Natale RB, Miller V, Manegold C, Scagliotti G, Rosell R, Oliff I, Reeves JA, Wolf MK, Krebs AD, Averbuch SD, Ochs JS, Grous J, Fandi A, Johnson DH (2004) Gefitinib in combination with paclitaxel and carboplatin in advanced non-small-cell lung cancer: a phase III trial-INTACT 2. J Clin Oncol 22: 785–794
Herbst RS, Prager D, Hermann R, Fehrenbacher L, Johnson BE, Sandler A, Kris MG, Tran HT, Klein P, Li X, Ramies D, Johnson DH, Miller VA, TRIBUTE Investigator Group (2005) TRIBUTE: a phase III trial of erlotinib hydrochloride (OSI-774) combined with carboplatin and paclitaxel chemotherapy in advanced non-small-cell lung cancer. J Clin Oncol 23: 5892–5899
Inoue A, Saijo Y, Maemondo M, Gomi K, Tokue Y, Kimura Y, Ebina M, Kikuchi T, Moriya T, Nukiwa T (2003) Severe acute interstitial pneumonia and gefitinib. Lancet 361: 137–139
Kaplan EL, Meier P (1958) Nonparametric estimation from incomplete observations. J Am Stat Assoc 53: 457–481
Kim KS, Jeong JY, Kim YC, Na KJ, Kim YH, Ahn SJ, Baek SM, Park CS, Park CM, Kim YI, Lim SC, Park KO (2005) Predictors of the response to gefitinib in refractory non-small cell lung cancer. Clin Cancer Res 11: 2244–2251
Kris MG, Natale RB, Herbst RS, Lynch Jr TJ, Prager D, Belani CP, Schiller JH, Kelly K, Spiridonidis H, Sandler A, Albain KS, Cella D, Wolf MK, Averbuch SD, Ochs JJ, Kay AC (2003) Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non-small cell lung cancer: a randomized trial. JAMA 290: 2149–2158
Kuroishi T, Hirose K, Tajima K (1999) Cancer mortality in Japan. In Gann Monograph Cancer Reserch Tominaga S, Ohshima A (eds) pp 1–3. Japan Scientific Societies Press: Tokyo
Lee DH, Han JY, Lee HG, Lee JJ, Lee EK, Kim HY, Kim HK, Hong EK, Lee JS (2005) Gefitinib as a first-line therapy of advanced or metastatic adenocarcinoma of the lung in never-smokers. Clin Cancer Res 11: 3032–3037
Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, Haber DA (2004) Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 350: 2129–2139
Mitsudomi T, Kosaka T, Endoh H, Horio Y, Hida T, Mori S, Hatooka S, Shinoda M, Takahashi T, Yatabe Y (2005) Mutations of the epidermal growth factor receptor gene predict prolonged survival after gefitinib treatment in patients with non-small-cell lung cancer with postoperative recurrence. J Clin Oncol 23: 2513–2520
Nagai Y, Miyazawa H, Huqun, Tanaka T, Udagawa K, Kato M, Fukuyama S, Yokote A, Kobayashi K, Kanazawa M, Hagiwara K (2005) Genetic heterogeneity of the epidermal growth factor receptor in non-small cell lung cancer cell lines revealed by a rapid and sensitive detection system, the peptide nucleic acid-locked nucleic acid PCR clamp. Cancer Res 65: 7276–7282
National Cancer Institute (1999) The revised common toxicity criteria: Version 2.0 CTEP Website http://ctep.info.nih.gov
Niho S, Kubota K, Goto K, Yoh K, Ohmatsu H, Kakinuma R, Saijo N, Nishiwaki Y (2006) First-line single agent treatment with gefitinib in patients with advanced non-small-cell lung cancer: a phase II study. J Clin Oncol 24: 64–69
Non-Small Cell Lung Cancer Collaborative Group (1995) Chemotherapy in non-small cell lung cancer – A meta analysis using updated data on individual patients from 52 randomized clinical trials. BMJ 311: 899–909
Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, Eck MJ, Sellers WR, Johnson BE, Meyerson M (2004) EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 304: 1497–1500
Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, Singh B, Heelan R, Rusch V, Fulton L, Mardis E, Kupfer D, Wilson R, Kris M, Varmus H (2004) EGF receptor gene mutations are common in lung cancers from ‘never smokers’ and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci USA 101: 13306–13311
Sasaki H, Endo K, Konishi A, Takada M, Kawahara M, Iuchi K, Matsumura A, Okumura M, Tanaka H, Kawaguchi T, Shimizu T, Takeuchi H, Yano M, Fukai I, Fujii Y (2005) EGFR Mutation status in Japanese lung cancer patients: genotyping analysis using LightCycler. Clin Cancer Res 11: 2924–2929
Scagliotti GV, Selvaggi G, Novello S, Hirsch FR (2004) The biology of epidermal growth factor receptor in lung cancer. Clin Cancer Res 10: 4227s–4232s
Schiller JH, Harrington D, Belani CP, Langer C, Sandler A, Krook J, Zhu J, Johnson DH, Eastern Cooperative Oncology Group (2002) Comparison of four chemotherapy regimens for advanced non-small-cell lung cancer. N Engl J Med 346: 92–98
Shepherd FA, Rodrigues Pereira J, Ciuleanu T, Tan EH, Hirsh V, Thongprasert S, Campos D, Maoleekoonpiroj S, Smylie M, Martins R, van Kooten M, Dediu M, Findlay B, Tu D, Johnston D, Bezjak A, Clark G, Santabarbara P, Seymour L, National Cancer Institute of Canada Clinical Trials Group (2005) Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med 353: 123–132
Simon R (1989) Optimal two-stage designs for phase II clinical trials. Control Clin Trials 10: 1–10
Takano T, Ohe Y, Sakamoto H, Tsuta K, Matsuno Y, Tateishi U, Yamamoto S, Nokihara H, Yamamoto N, Sekine I, Kunitoh H, Shibata T, Sakiyama T, Yoshida T, Tamura T (2005) Epidermal growth factor receptor gene mutations and increased copy numbers predict gefitinb sensitivity in patients with recurrent non-small-cell lung cancer. J Clin Oncol 23: 6829–6837
Taron M, Ichinose Y, Rosell R, Mok T, Massuti B, Zamora L, Mate JL, Manegold C, Ono M, Queralt C, Jahan T, Sanchez JJ, Sanchez-Ronco M, Hsue V, Jablons D, Sanchez JM, Moran T (2005) Activating mutations in the tyrosine kinase domain of the epidermal growth factor receptor are associated with improved survival in gefitinib-treated chemorefractory lung adenocarcinomas. Clin Cancer Res 11: 5878–5885
Thatcher N, Chang A, Parikh P, Rodrigues Pereira J, Ciuleanu T, von Pawel J, Thongprasert S, Tan EH, Pemberton K, Archer V, Carroll K (2005) Gefitinib plus best supportive care in previously treated patients with refractory advanced non-small-cell lung cancer: results from a randomised, placebo-controlled, multicentre study (Iressa Survival Evaluation in Lung Cancer). Lancet 366: 1527–1537
Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG (2000) New guidelines to evaluate the response to treatment in solid tumors. J Natl Cancer Inst 92: 205–216
Tsao MS, Sakurada A, Cutz JC, Zhu CQ, Kamel-Reid S, Squire J, Lorimer I, Zhang T, Liu N, Daneshmand M, Marrano P, da Cunha Santos G, Lagarde A, Richardson F, Seymour L, Whitehead M, Ding K, Pater J, Shepherd FA (2005) Erlotinib in lung cancer – molecular and clinical predictors of outcome. New Engl J Med 353: 133–144
Zhang XT, Li LY, Mu XL, Cui QC, Chang XY, Song W, Wang SL, Wang MZ, Zhong W, Zhang L (2005) The EGFR mutation and its correlation with response of gefitinib in previously treated Chinese patients with advanced non-small-cell lung cancer. Ann Oncol 16: 1334–1342
Author information
Authors and Affiliations
Consortia
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Asahina, H., Yamazaki, K., Kinoshita, I. et al. A phase II trial of gefitinib as first-line therapy for advanced non-small cell lung cancer with epidermal growth factor receptor mutations. Br J Cancer 95, 998–1004 (2006). https://doi.org/10.1038/sj.bjc.6603393
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6603393
Keywords
This article is cited by
-
Differential efficacy of tyrosine kinase inhibitors according to the types of EGFR mutations and agents in non-small cell lung cancer: a real-world study
BMC Cancer (2024)
-
Empirical methods for the validation of time-to-event mathematical models taking into account uncertainty and variability: application to EGFR + lung adenocarcinoma
BMC Bioinformatics (2023)
-
Knowledge-based mechanistic modeling accurately predicts disease progression with gefitinib in EGFR-mutant lung adenocarcinoma
npj Systems Biology and Applications (2023)
-
Third-generation EGFR and ALK inhibitors: mechanisms of resistance and management
Nature Reviews Clinical Oncology (2022)
-
The emerging treatment landscape of targeted therapy in non-small-cell lung cancer
Signal Transduction and Targeted Therapy (2019)
