
Abstract
KIT is a tyrosine kinase receptor expressed by several tumours, which has for specific ligand the stem cell factor (SCF). KIT is the main oncogene in gastrointestinal stromal tumours (GISTs), and gain-of-function KIT mutations are present in 70% of these tumours. The aim of the study was to measure and investigate the mechanisms of KIT activation in 80 KIT-positive GIST patients. KIT activation was quantified by detecting phosphotyrosine residues in Western blotting. SCF production was determined by reverse transcriptase–PCR, ELISA and/or immunohistochemistry. Primary cultures established from three GISTs were also analysed. The results show that KIT activation was detected in all cases, even in absence of KIT mutations. The fraction of activated KIT was not correlated with the mutational status of GISTs. Membrane and soluble isoforms of SCF mRNA were present in all GISTs analysed. Additionally, SCF was also detected in up to 93% of GISTs, and seen to be present within GIST cells. Likewise, the two SCF mRNA isoforms were found to be expressed in GIST-derived primary cultures. Thus, KIT activation in GISTs may in part result from the presence of SCF within the tumours.
Similar content being viewed by others

Main
Proto-oncogene KIT is a class III transmembrane tyrosine kinase receptor (TKR) (Reilly, 2003) which is involved in the growth and differentiation of haematopoietic stem cells, mast cells, melanocytes, germinal cells and Cajal's interstitial cells (Ashman, 1999). The binding of KIT ligand promotes KIT dimerization, and activates its intrinsic tyrosine kinase activity, thereby resulting in a transphosphorylation at several critical tyrosine residues and an activation of downstream signal transduction molecules (Blume-Jensen et al, 1991). The ligand for KIT receptor is the stem cell factor (SCF) also known as KIT ligand, steel factor and mast cell growth factor. SCF is implicated in cell proliferation (Williams and Allan, 1996; Drayer et al, 2005), migration and survival (Bredin et al, 2003; Erlandsson et al, 2004). Recently, SCF has also been shown to be implicated in a resistance mechanism of malignant mesothelioma cells to drug treatment (Catalano et al, 2004). SCF expression is widely distributed throughout the body, especially in stromal cells such as fibroblasts and endothelial cells, and detectable at low levels in the blood (Ashman, 1999). SCF exists under soluble (s) and membrane-bound (m) forms due to differential splicing and proteolytic cleavage (Broudy, 1997). The two forms display distinct effects as regards to the survival and proliferation of haematopoietic cell lines (Caruana et al, 1993; Miyazawa et al, 1995) and primary cells (Fujita et al, 1989; Toksoz et al, 1992), although they are both active in increasing the number of human progenitor cells in the context of stromal cell cultures (Toksoz et al, 1992). Stromal mSCF appears to induce more persistent signalling than the soluble form, this last form inducing rapid downregulation of cell surface expression and degradation of KIT (Miyazawa et al, 1995).
KIT is detectable in several human tumours (Went et al, 2004), and paracrine/autocrine activation by its ligand has been suggested to be involved in numerous malignancies, including small-cell lung cancer (Hibi et al, 1991; Krystal et al, 1996), ovarian cancer (Inoue et al, 1994), neuroblastoma (Ricotti et al, 1998), breast carcinoma (Hines et al, 1995), leukaemia (Pietsch, 1993; Zheng et al, 2004), colon carcinoma (Lahm et al, 1995) and Leydig cell tumour (Kondoh et al, 1995). Gastrointestinal stromal tumours (GISTs) are the most frequent mesenchymal tumours of the digestive tract. Nearly all of them express KIT and 70% have a gain-of-function mutation of KIT gene (Corless et al, 2004), responsible for ligand-independent KIT activation (Hirota et al, 1998). Moreover, rare GISTs have been shown to be related to familial KIT mutations (Nishida et al, 1998), and transgenic mice with a gain-of-function mutation of KIT have a high incidence of GIST (Sommer et al, 2003). Finally, treatment of GIST patients with the KIT inhibitor Imatinib can induce tumour regression in many patients (Heinrich et al, 2003a). Thus, KIT activation plays a major role in GIST oncogenesis. Surprisingly, there are a few data dealing with SCF in GISTs and its possible role as an autocrine/paracrine growth factor.
In the present study, we quantified the activation of KIT in GIST samples and found that there is no correlation with the presence of gain-of-function mutations. We also show that the two isoforms of the KIT ligand SCF are present in nearly all GISTs, and are produced by tumour cells.
Materials and methods
Patients
A total of 80 patients with GIST were included in the study. For all cases, GIST diagnosis was confirmed by at least two pathologists. Haematoxyline–eosin–saffran staining and immunohistochemistry were performed with CD117/KIT (rabbit polyclonal, Dako, Copenhagen, Denmark, 1 : 300 dilution), CD34 (mouse, clone QBEND, Immunotech, Marseille, France, 1 : 2 dilution), and S100 protein (rabbit polyclonal, Dako, 1 : 200 dilution). Detection of exon 9, 11, 13, 17 mutations of KIT, and exon 12 and 18 mutations of PDGFRA was performed on either formalin fixed or frozen tumour samples as previously described (Emile et al, 2002, 2004). Clinical and pathological characteristics, as well as mutational status of most cases, have been previously reported (Emile et al, 2004; Théou et al, 2004; Tabone et al, 2005). Frozen and fresh tumour samples were obtained from 18 and three patients, respectively. Tumour samples were collected during surgical resection performed for therapeutic purposes and harvested before treatment with imatinib mesylate. According to French ethical laws, none of the patients expressed his willingness not to be included in this study. Paraffin-embedded and frozen normal digestive tissue samples from eleven patients without GIST were used as controls. None of them displayed mutations in KIT or PDGFRA.
Cell isolation and primary cell culture
Fresh GIST samples were minced with scissors, washed twice in PBS, and incubated in a 0.25% solution of collagenase A (Roche Diagnostics, Meylan, France) at +37°C until disaggregation was complete. After cold centrifugation in PBS, cell pellets were resuspended in 1640 RPMI medium supplemented with 10%s FCS (both from Gibco BRL, Cergy-Pontoise, France), and seeded into culture flasks (ATGC Biotechnologie, Noisy-le-Grand, France) at approximately 1.0 × 105 cells cm−2. Medium changes were performed 24 h after seeding, and once a week before cell analysis. For RNA extraction, confluent cultures were trypsinized, and cells were centrifuged at 2000 r.p.m. for 5 min at room temperature. Cell pellets were vortexed in lysis buffer (350 μl/106 cells) of RNeasy KIT for RNA extraction (Qiagen, Courtaboeuf, France), and kept at −80°C before analysis.
Detection of soluble and membrane-bound forms of SCF mRNA
For RNA extraction, frozen tissues were mechanically homogenized (Mixer Mill MM 300, Retsch, Germany). RNA was extracted from homogenates of tumours, digestive tissues and cultured cells with the RNeasy & QIAshredder KITs (Qiagen) according to the manufacturer's instructions. A maximum of 1 μg of RNA was reverse transcribed in a 20 μl final volume. The reaction mixture contained 1 × RT buffer, 5.5 mM MgCl2, 500 μ M each dNTP, 2.5 μM random hexamers, 0.4 U μl−1 RNase inhibitor and 1.25 U μl−1 reverse transcriptase (Applied Biosystems, Foster City, CA, USA). The cycling conditions were 10 min at 25°C, 30 min at 48°C, and 5 min at 95°C. Soluble and membrane-bound SCF mRNA were detected by PCR amplification using primers SCF-F-HEX (5′CAAGGACTTTGTAGTGGCATCT3′) and SCF-R (5′GAGAAAACAATGCTGGCAAT3′) on RT–PCR products. Each of the 35 PCR cycles consisted of denaturation for 1 min at 92°C, annealing for 30 s at 60°C and elongation for 45 s at 72°C. Each PCR included an initial denaturation step of 2 min at 94°C and a final elongation step of 7 min at 72°C. To detect the PCR HEX-labelled products and to determine their size, 1 μl of each sample was added to 23 μl of formamide and 0.5 μl of GeneScan 500 TAMRA size standard (Applied Biosystems, Foster City, CA, USA). Capillary electrophoresis was performed with an ABI Prism 310 apparatus, according to the manufacturer's instructions. Size curves and fluorescence intensity were analysed and quantified with Genescan software (Applied Biosystems).
Immunohistochemical detection of SCF
Paraffin-embedded donor tissue blocks were sampled with 0.6 punchers using a tissue microarray (TMA) instrument (Beecher Instruments Inc., Sun Prairie, WI, USA). Previously haematoxylin–eosin–saffran stained sections were used to select representative tumour areas, and for each tumour, three cylindrical cores were removed from the donor block and placed into the recipient TMA paraffin block. After antigen removal, TMA sections were subjected to immunohistochemical staining by using anti-KIT (1 : 300 dilution), DOG-1 (rabbit polyclonal S0284, Applied Genomics Inc., Huntsville, AL, USA, 1 : 40 dilution) and SCF (rabbit polyclonal H-189, Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA, 1 : 40 dilution) antibodies, and avidin–biotin complex immunoperoxidase technique (LSAB2, Dako). Immunohistochemistry was also performed on 10 large GIST samples, which contained tumour as well as adjacent nontumour tissue.
Protein extraction and analysis
Frozen GIST samples were calibrated and mechanically homogenized (Mixer Mill MM 300) in lysis buffer (20 mM Tris, 150 mM NaCl, 1 mM othovanadate, 10 mM NaF, 1 mM PMSF, 0.5 μg ml−1 leupeptine, 1 μg ml−1 pepstatine, 10 KIU ml−1 aprotinine, 1% Triton X-100). Lysates were rocked for 30 min at 4°C, and then centrifuged to remove insoluble material. Supernatant protein contents were determined using Bradford solution and were normalized.
KIT was immunoprecipitated from 100 μl of lysates, using anti-CD117 antibody and Sepharose protein G beads (Amersham Pharmacia Biotech, Les Ulis, France). After 1 h incubation at 4°C under constant agitation, the beads were washed three times in lysis buffer. Immunoprecipitates and total lysates were resuspended in Laemmli buffer, heated and separated by 5–15% SDS–polyacrylamide gel electrophoresis under reducing conditions, and transferred to a polyvinylidene difluoride membrane (Biorad, Marnes-la Coquette, France). Western blots on immuno-precipitates were performed using 4G10 (kindly provided by Dr C Boucheix, 1 : 5 dilution) phosphotyrosine mAb and anti-CD117 (rabbit polyclonal, Dako, 1 : 1000 dilution) antibody whereas Western blots on total lysates was performed using TYR 721 Antibody (Voytyuk et al, 2003, rabbit polyclonal, 1 μg ml−1)). Immunoreactive bands were visualized by using appropriate secondary horseradish peroxidase-conjugated antibodies (Immunotech, Marseille, France) and enhanced chemiluminescence (PerkinElmer, Life Sciences). Measurement of fluorescent intensity was performed by using Intelligent Dark box II (Fujifilm, FUJI, Japan) and Image Gauge V4.0 5F (Fujifilm) sofware. Quantitative determinations of human SCF concentrations were performed using 100 μl of protein extracts by ELISA (Quantikine, R&D systems, USA) according to the manufacturer's instructions.
Results
KIT activation in GISTs
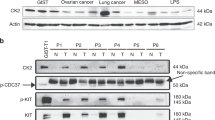
KIT activation was determined in total protein extracts prepared from 17 frozen GIST samples by two different methods. In the first step, analyses was performed by Western blotting using TYR721 antibody, specific for the phosphorylated tyrosine 721. A positive signal was detected in 14/17 cases (Figure 1). However, the total amount of activated KIT highly varied between tumours. To quantify the fraction of activated KIT present in each GIST sample, total protein extracts were immunoprecipitated with anti-KIT antibody. After Western blotting, dilutions were made to obtain the same amount of KIT in each sample. KIT activation was then analysed on a second Western blot using 4G10 mAb, specific for phosphotyrosine. As illustrated in Figure 2, a positive signal was detected at 145 kDa in all cases and the fraction of KIT activation differed between samples. The fraction of activated KIT revealed by 4G10 mAb was then quantified in GIST samples (Table 1), no correlation was found with the presence of KIT mutations (P>0.05, nonparametric Wilcoxon test).

Expression of phosphorylated Tyrosine 721 of KIT protein. For each sample, 18 μg of total protein extracts were loaded in gels and revealed on Western blot by Y721 polyclonal antibody specific for the phosphorylated tyrosine 721 (upper gel) or polyclonal anti-KIT antibody (lower gel). Wild type GIST: A (71105) and B (71183). Mutated GIST C (71229), D (71175), E (71100), F (71237), G (70810) and H (71101).

Expression of phosphorylated KIT protein. Protein extracts were immunoprecipitated by KIT antibody. A first Western blot was revealed with KIT antibody to quantify the amount of KIT in each samples. Appropriate dilutions were then performed to load the same amount of KIT from each samples in a second gel revealed by 4G10 mAb (upper gel) and a third gel revealed by KIT (lower gel). On the upper gel, the bands of 145 kDa corresponded to activated KIT protein. The lower gel confirmed that similar amounts of total KIT were loaded from each samples. Wild-type GIST: A (71186) and B (71105). Mutated GIST C (71237), D (71101), E (70810), F (71231) and G (71562).
Detection of SCF in GISTs
As KIT activation did not depend on the gain-of-function mutations, we looked for the presence of the KIT ligand SCF within the tumours. Indeed, SCF mRNAs were detected in the 18 frozen GISTs samples tested by RT–PCR. SCF was also detected in protein extracts from eight GISTs by ELISA with concentrations ranging from 3.7 to 89 pg ml−1 of total protein extract (mean 29.6 pg ml−1). Interestingly, the lowest SCF amount was found in a tumour in which PDGFRA mutation had been detected. To further confirm the presence of SCF in GISTs, we performed immunohistochemistry on tissue microarray containing 67 GISTs positive for both KIT and DOG-1. In all, 62 cases (93%) were positive for SCF.
As seen in Figure 3, primers for RT-PCR allowed amplifying the two SCF isoforms. The larger amplicon (222 base pairs) corresponded to the sSCF, containing a proteolytic domain. The smaller amplicon (137 base pair) did no contain this domain and corresponded to the mSCF. sSCF signal was higher in most (14/18) cases.

Quantification of SCF isoforms by fluorescence intensity after RT–PCR in GISTs and digestive tissues. Fluorescent amplicons (blue peaks) were separated by capillary electrophoresis and identified on the basis of their size. Peaks at 222 base pairs (arrow head) corresponded to sSCF, while peaks at 137 base pairs (arrow) corresponded to mSCF. The green peaks corresponded to size markers (TAMRA). Numbers 71237 and 70810 are GISTs samples, and 71387 and 71388 are digestive tissues from muscularis propria (ileon) and mucosa (colon), respectively.
Production of SCF by tumour and nontumour cells
SCF is known to be produced by several types of cells. Indeed, SCF mRNA was detected in the 11 normal digestive control samples that we tested (Table 1). SCF was also detected by ELISA in two of three control digestive samples.
Immunohistochemistry analysis showed that, in the 63 GIST samples positive for SCF, the staining was mainly detected within tumour cells. In nearly all positive cases, cellular staining was diffuse, and generally displayed a membranous and cytoplasmic distribution pattern (Figure 4).

Detection of SCF staining within GIST cells by immunohistochemistry with anti-SCF antibody. A positive diffuse staining was present in most GISTs (A–C), but absent in some cases (D). The staining was generally both cytoplasmic and membranous (A), and sometimes predominantly membranous (B) or cytoplasmic (C).
To further confirm that GIST cells were able to produce SCF, primary cultures were established from three fresh tumours derived from the stomach, and all strongly positive for KIT. Primary cultures consisted of a homogeneous population of spindle-shaped cells (Figure 5). The heterozygous KIT exon 11 deletions detected in two out of the three tumours, were similarly present in the corresponding primary cultures. Moreover, RT–PCR also revealed the presence of both both sSCF and mSCF.

Primary cultures of GIST cells. Primary cultures of GISTs showed a homogeneous population of spindle-shaped cells (primary culture of GIST 70810, original magnification × 400).
Immunohistochemistry performed on larger histological samples, which contained GIST as well as nontumour adjacent tissue, confirmed the diffuse staining within GIST and also showed a positivity in some nontumour cells. Staining of muscular cells of the muscularis propria and of the arteries was always lower than that of GIST cells (Figure 6). By contrast, some lymphocytes had a higher positivity (Figure 6).

Detection of SCF in GISTs and nontumour cells. Immunohistochemistry was performed on large GIST samples with anti-SCF. (A) The intensity of staining of the muscularis propria (arrows) was lower than that of GIST. (B) Within GISTs, some lymphocytes had a strong staining (arrows), while arterial walls were almost negative (arrow heads).
Discussion
KIT plays a major role in GIST oncogenesis. Indeed, 95% of GISTs express KIT (Corless et al, 2004), and its inhibitor, Imatinib, shows strong antitumour effects (Demetri et al, 2002). About 70% of GISTs display gain-of-function mutation of KIT (Corless et al, 2004) responsible for constitutive KIT activation (Hirota et al, 1998). These mutations also play a major role in GIST oncogenesis. Indeed, they are associated with some familial cases (Nishida et al, 1998), and similarly KIT-mutated knock-in mice display high GIST incidence (Sommer et al, 2003). However, most data dealing with KIT activation by gain-of-function mutations are obtained in cells containing a homozygous mutation, while 94% of GISTs' mutations are heterozygous (Emile et al, 2004). It is, thus, not yet known whether heterozygous KIT mutations act by inducing overall KIT activation or by other mechanisms, such as activation of specific signal transduction pathways in GIST cells.
Our analysis of the activation status of KIT in 17 KIT-positive GISTs by the detection of phosphorylated tyrosine residues shows that at least a fraction of KIT was activated in each tumour, even in the absence of gain-of-function mutation, consistent with previously published data (Rubin et al, 2001; Heinrich et al, 2003b; Antonescu et al, 2005). After immunoprecipitation and adjustment of the total KIT amounts in each sample, we were able to quantify the activated fraction of KIT. In these conditions, we found no correlation with the mutational GIST's status. Thus, the heterozygous gain-of-function mutations of KIT in GISTs have a few impact on the overall activation of this tyrosine kinase receptor. These results prompted us to look for an alternative mechanism of KIT activation in GISTs.
The absence of correlation between mutational status and KIT activation could be explained by the presence of SCF within tumours. Indeed, we show that SCF transcripts were present in the entire GIST samples tested, results consistent with a recently published cDNA array study (Antonescu et al, 2004). We also detected SCF by ELISA and immunohistochemistry in up to 93% of GIST samples. SCF treatment of GIST544 cells, which express a heterozygous KIT exon 9 mutation induces a stronger KIT tyrosine phosphorylation (Duensing et al, 2004), while SCF treatment of GIST882 cells, which carry a homozygous KIT exon 13 mutation does not (Lux et al, 2000). The two main isoforms of KIT, GNNK− and GNNK+, have been shown to have differential biological activities. Indeed GNNK−, which is the most abundant in GISTs (Théou et al, 2004), strongly promotes colony formation in semisolid medium, loss of cell to cell contact inhibition, and tumour growth in nude mice (Caruana et al, 1999). Furthermore, after ligand stimulation, the GNNK− isoform displayed more rapid and extensive tyrosine autophosphorylation and faster internalization (Caruana et al, 1999). Thus SCF may activate the wild-type species of KIT in GISTs without mutations, and may also modulate the fraction of activated KIT in GISTs with heterozygous mutations.
The presence of both KIT and SCF has already been reported in several tumours (Hibi et al, 1991; Pietsch, 1993; Inoue et al, 1994; Hines et al, 1995; Kondoh et al, 1995; Lahm et al, 1995; Krystal et al, 1996; Ricotti et al, 1998; Zheng et al, 2004), suggesting either an autocrine or paracrine oncogenic effect. The cellular origin of SCF in GIST samples appears to be the tumour cells themselves. Immunohistochemistry disclosed a strong staining of most GIST cells with anti-SCF antibody. This staining, however, could result from the internalization of SCF after its ligation to KIT. Indeed, SCF is known to be produced by different cell types, and we did detect SCF mRNA in all the nontumoural digestive samples tested. This result highlights that microenvironnement of tumour cells could led to a paracrine loop mechanism. However, by establishing primary homogenous GIST cell cultures, we confirmed the SCF production, both membrane and soluble isoforms of SCF mRNA, in GIST cells.
In conclusion, although KIT gain-of-function mutations play a major role in GIST oncogenesis, we show that KIT activation in these tumours is unrelated to the presence of these mutations, and may result from an autocrine/paracrine mechanism. Therefore, heterozygous KIT mutations in GISTs may act by inducing specific signal transduction pathways, rather than enhancing overall KIT activation.
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Antonescu CR, Besmer P, Guo T, Arkun K, Hom G, Koryotowski B, Leversha MA, Jeffrey PD, Desantis D, Singer S, Brennan MF, Maki RG, DeMatteo RP (2005) Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clin Cancer Res 11 (11): 4182–4190
Antonescu CR, Viale A, Sarran L, Tschernyavsky SJ, Gonen M, Segal NH, Maki RG, Socci ND, DeMatteo RP, Besmer P (2004) Gene expression in gastrointestinal stromal tumors is distinguished by KIT genotype and anatomic site. Clin Cancer Res 10 (10): 3282–3290
Ashman LK (1999) The biology of stem cell factor and its receptor C-KIT. Int J Biochem Cell Biol 31 (10): 1037–1051
Blume-Jensen P, Claesson-Welsh L, Siegbahn A, Zsebo KM, Westermark B, Heldin CH (1991) Activation of the human c-KIT product by ligand-induced dimerization mediates circular actin reorganization and chemotaxis. EMBO J 10 (13): 4121–4128
Bredin C, Liu GZ, Klominek J (2003) Growth factor-enhanced expression and activity of matrix metalloproteases in human non-small cell lung cancer cell lines. Anticancer Res 23 (6C): 4877–4884
Broudy VC (1997) Stem cell factor and hematopoiesis. Blood 90 (4): 1345–1364
Caruana G, Ashman LK, Fujita J, Gonda TJ (1993) Responses of the murine myeloid cell line FDC-P1 to soluble and membrane-bound forms of steel factor (SLF). Exp Hematol 21 (6): 761–768
Caruana G, Cambareri AC, Ashman LK (1999) Isoforms of c-KIT differ in activation of signalling pathways and transformation of NIH3T3 fibroblasts. Oncogene 18 (40): 5573–5581
Catalano A, Rodilossi S, Rippo MR, Caprari P, Procopio A (2004) Induction of stem cell factor/c-KIT/slug signal transduction in multidrug-resistant malignant mesothelioma cells. J Biol Chem 279 (45): 46706–46714
Corless CL, Fletcher JA, Heinrich MC (2004) Biology of gastrointestinal stromal tumors. J Clin Oncol 22 (18): 3813–3825
Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, Heinrich MC, Tuveson DA, Singer S, Janicek M, Fletcher JA, Silverman SG, Silberman SL, Capdeville R, Kiese B, Peng B, Dimitrijevic S, Druker BJ, Corless C, Fletcher CD, Joensuu H (2002) Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med 347 (7): 472–480
Drayer AL, Boer AK, Los EL, Esselink MT, Vellenga E (2005) Stem cell factor synergistically enhances thrombopoietin-induced STAT5 signaling in megakaryocyte progenitors through JAK2 and Src kinase. Stem Cells 23 (2): 240–251
Duensing A, Medeiros F, McConarty B, Joseph NE, Panigrahy D, Singer S, Fletcher CD, Demetri GD, Fletcher JA (2004) Mechanisms of oncogenic KIT signal transduction in primary gastrointestinal stromal tumors (GISTs). Oncogene 23 (22): 3999–4006
Emile JF, Lemoine A, Bienfait N, Terrier P, Azoulay D, Debuire B (2002) Length analysis of polymerase chain reaction products: a sensitive and reliable technique for the detection of mutations in KIT exon 11 in gastrointestinal stromal tumors. Diagn Mol Pathol 11 (2): 107–112
Emile JF, Theou N, Tabone S, Cortez A, Terrier P, Chaumette MT, Julie C, Bertheau P, Lavergne-Slove A, Donadieu J, Barrier A, Le Cesne A, Debuire B, Lemoine A, Groupe d'Etude des GIST (2004) Clinicopathologic, phenotypic, and genotypic characteristics of gastrointestinal mesenchymal tumors. Clin Gastroenterol Hepatol 2 (7): 597–605
Erlandsson A, Larsson J, Forsberg-Nilsson K (2004) Stem cell factor is a chemoattractant and a survival factor for CNS stem cells. Exp Cell Res 301 (2): 201–210
Fujita J, Onoue H, Ebi Y, Nakayama H, Kanakura Y (1989) In vitro duplication and in vivo cure of mast-cell deficiency of Sl/Sld mutant mice by cloned 3T3 fibroblasts. Proc Natl Acad Sci USA 86 (8): 2888–2891
Heinrich MC, Corless CL, Demetri GD, Blanke CD, von Mehren M, Joensuu H, McGreevey LS, Chen CJ, Van den Abbeele AD, Druker BJ, Kiese B, Eisenberg B, Roberts PJ, Singer S, Fletcher CD, Silberman S, Dimitrijevic S, Fletcher JA (2003a) Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol 21 (23): 4342–4349
Heinrich MC, Corless CL, Duensing A, McGreevey L, Chen CJ, Joseph N, Singer S, Griffith DJ, Haley A, Town A, Demetri GD, Fletcher CD, Fletcher JA (2003b) PDGFRA activating mutations in gastrointestinal stromal tumors. Science 299 (5607): 708–710
Hibi K, Takahashi T, Sekido Y, Ueda R, Hida T, Ariyoshi Y, Takagi H, Takahashi T (1991) Coexpression of the stem cell factor and the c-KIT genes in small-cell lung cancer. Oncogene 6 (12): 2291–2296
Hines SJ, Organ C, Kornstein MJ, Krystal GW (1995) Coexpression of the c-KIT and stem cell factor genes in breast carcinomas. Cell Growth Differ 6 (6): 769–779
Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, Kawano K, Hanada M, Kurata A, Takeda M, Muhammad Tunio G, Matsuzawa Y, Kanakura Y, Shinomura Y, Kitamura Y (1998) Gain-of-function mutations of c-KIT in human gastrointestinal stromal tumors. Science 279 (5350): 577–580
Inoue M, Kyo S, Fujita M, Enomoto T, Kondoh G (1994) Coexpression of the c-KIT receptor and the stem cell factor in gynecological tumors. Cancer Res 54 (11): 3049–3053
Kondoh G, Hayasaka N, Li Q, Nishimune Y, Hakura A (1995) An in vivo model for receptor tyrosine kinase autocrine/paracrine activation: auto-stimulated KIT receptor acts as a tumor promoting factor in papillomavirus-induced tumorigenesis. Oncogene 10 (2): 341–347
Krystal GW, Hines SJ, Organ CP (1996) Autocrine growth of small cell lung cancer mediated by coexpression of c-KIT and stem cell factor. Cancer Res 56 (2): 370–376
Lahm H, Amstad P, Yilmaz A, Borbenyi Z, Wyniger J, Fischer JR, Suardet L, Givel JC, Odartchenko N (1995) Interleukin 4 down-regulates expression of c-KIT and autocrine stem cell factor in human colorectal carcinoma cells. Cell Growth Differ 6 (9): 1111–1118
Lux ML, Rubin BP, Biase TL, Chen CJ, Maclure T, Demetri G, Xiao S, Singer S, Fletcher CD, Fletcher JA (2000) KIT extracellular and kinase domain mutations in gastrointestinal stromal tumors. Am J Pathol 156 (3): 791–795
Miyazawa K, Williams DA, Gotoh A, Nishimaki J, Broxmeyer HE, Toyama K (1995) Membrane-bound Steel factor induces more persistent tyrosine kinase activation and longer life span of c-KIT gene-encoded protein than its soluble form. Blood 85 (3): 641–649
Nishida T, Hirota S, Taniguchi M, Hashimoto K, Isozaki K, Nakamura H, Kanakura Y, Tanaka T, Takabayashi A, Matsuda H, Kitamura Y (1998) Familial gastrointestinal stromal tumours with germline mutation of the KIT gene. Nat Genet 19 (4): 323–324
Pietsch T (1993) Paracrine and autocrine growth mechanisms of human stem cell factor (c-KIT ligand) in myeloid leukemia. Nouv Rev Fr Hematol 35 (3): 285–286
Reilly JT (2003) Receptor tyrosine kinases in normal and malignant haematopoiesis. Blood Rev 17 (4): 241–248
Ricotti E, Fagioli F, Garelli E, Linari C, Crescenzio N, Horenstein AL, Pistamiglio P, Vai S, Berger M, di Montezemolo LC, Madon E, Basso G (1998) c-KIT is expressed in soft tissue sarcoma of neuroectodermic origin and its ligand prevents apoptosis of neoplastic cells. Blood 91 (7): 2397–2405
Rubin BP, Singer S, Tsao C, Duensing A, Lux ML, Ruiz R, Hibbard MK, Chen CJ, Xiao S, Tuveson DA, Demetri GD, Fletcher CD, Fletcher JA (2001) KIT activation is a ubiquitous feature of gastrointestinal stromal tumors. Cancer Res 61 (22): 8118–8121
Sommer G, Agosti V, Ehlers I, Rossi F, Corbacioglu S, Farkas J, Moore M, Manova K, Antonescu CR, Besmer P (2003) Gastrointestinal stromal tumors in a mouse model by targeted mutation of the KIT receptor tyrosine kinase. Proc Natl Acad Sci USA 100 (11): 6706–6711
Tabone S, Theou N, Wozniak A, Saffroy R, Deville L, Julie C, Callard P, Lavergne-Slove A, Debiec-Rychter M, Lemoine A, Emile JF (2005) KIT overexpression and amplification in gastrointestinal stromal tumors (GISTs). Biochim Biophys Acta 1741 (1–2): 165–172
Théou N, Tabone S, Saffroy R, Le Cesne A, Julie C, Cortez A, Lavergne-Slove A, Debuire B, Lemoine A, Emile JF (2004) High expression of both mutant and wild-type alleles of c-KIT in gastrointestinal stromal tumors. Biochim Biophys Acta 1688 (3): 250–256
Toksoz D, Zsebo KM, Smith KA, Hu S, Brankow D, Suggs SV, Martin FH, Williams DA (1992) Support of human hematopoiesis in long-term bone marrow cultures by murine stromal cells selectively expressing the membrane-bound and secreted forms of the human homolog of the steel gene product, stem cell factor. Proc Natl Acad Sci USA 89 (16): 7350–7354
Voytyuk O, Lennartsson J, Mogi A, Caruana G, Courtneidge S, Ashman LK, Ronnstrand L (2003) Src family kinases are involved in the differential signaling from two splice forms of c-KIT. J Biol Chem 278 (11): 9159–9166
Went PT, Dirnhofer S, Bundi M, Mirlacher M, Schraml P, Mangialaio S, Dimitrijevic S, Kononen J, Lugli A, Simon R, Sauter G (2004) Prevalence of KIT expression in human tumors. J Clin Oncol 22 (22): 4514–4522
Williams B, Allan DJ (1996) Combination of SCF, IL-6, IL-3, and GM-CSF increases the mitotic index in short term bone marrow cultures from acute promyelocytic leukemia (APL) patients. Cancer Genet Cytogenet 91 (1): 77–81
Zheng R, Klang K, Gorin NC, Small D (2004) Lack of KIT or FMS internal tandem duplications but co-expression with ligands in AML. Leuk Res 28 (2): 121–126
Acknowledgements
The authors thank C Boucheix, M-C Le Bousse-Kerdilès, E Rubinstein, and F Chiappini for fruitful discussions; C Julié, A Cortez, and A Lavergne-Slove for providing frozen GIST samples; Y Pothin, D Péchaud and M Bakhari for excellent technical assistance. This work was supported by grants from the PHRC AOR03055, BQR2004 Faculté de Médecine Paris Ile de France Ouest and GRIP. S Tabone is a fellow of ANRT, with financial support of Léon Bérard Cancer Center and Novartis SA.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Théou-Anton, N., Tabone, S., Brouty-Boyé, D. et al. Co expression of SCF and KIT in gastrointestinal stromal tumours (GISTs) suggests an autocrine/paracrine mechanism. Br J Cancer 94, 1180–1185 (2006). https://doi.org/10.1038/sj.bjc.6603063
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6603063
Keywords
This article is cited by
-
SPRY4 inhibits and sensitizes the primary KIT mutants in gastrointestinal stromal tumors (GISTs) to imatinib
Gastric Cancer (2023)
-
Proteomic detection of a large amount of SCGFα in the stroma of GISTs after imatinib therapy
Journal of Translational Medicine (2011)
-
Silent mutations in KIT and PDGFRA and coexpression of receptors with SCF and PDGFA in Merkel cell carcinoma: implications for tyrosine kinase-based tumorigenesis
Modern Pathology (2008)
